

Downloads
Download



This work is licensed under a Creative Commons Attribution 4.0 International License.
Review
Promising Treatments for Duchenne Muscular Dystrophy: Restoring Dystrophin Protein Expression Using Nucleic Acid Therapeutics
Guo Hu and Chen Chen *
Division of Cardiology and Hubei Key Laboratory of Genetics and Molecular Mechanisms of Cardiological Disorders, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
* Correspondence: chenchen@tjh.tjmu.edu.cn; Tel. & Fax: 86-27-6937-8422
Received: 10 October 2022
Accepted: 10 November 2022
Published: 11 January 2023
Abstract: Duchenne muscular dystrophy is caused by inadequate generation of functional dystrophin protein. Traditional clinical treatments can only slightly mitigate the progression of the disease, but not completely stem or reverse the decline in muscle function. Therapies aimed at dystrophin recovery are currently under development, among which are exon skipping and stop codon readthrough therapies. They are now used in clinics, while gene addition therapies are in phase III clinical trials. Gene editing therapies have also been approved for the first clinical trial recently. This review will discuss these emerging therapies, clinical trials, and directions for future developments.
Keywords:
duchenne muscular dystrophy dystrophin nucleic acid therapeutics1. Introduction
Duchenne muscular dystrophy (DMD) is a severe progressive X-chromosome genetic disorder that affects approximately 19.8 out of every 100,000 male infants worldwide [1]. Boys with DMD typically begin to exhibit symptoms after the first year of life. Early symptoms include frequent falls, inability to run and climb stairs. By the age of twelve, many experience complete loss of independent walking ability, followed by the gradual development of cardiomyopathy and respiratory failure, and finally early death due to respiratory failure [2].
The dystrophin gene has 79 exons to encode dystrophin protein. Frameshift and nonsense mutations in the dystrophin gene might result in DMD; these mutations lead to premature termination of translation and production of non-functional dystrophin. Dystrophin protein has four structural domains: the N-terminal domain; the rod domain; the cysteine-rich domain (CR); the C-terminal domain (CT) [4]. Together with sarcoglycans, sarcospan, dystrobrevin, syntrophins and neuronal nitric oxide synthase (nNOS), they constitute a major part of the dystrophin-associated protein complex (DAPC). DAPC acts as a membrane stabilizer during myocyte contraction [5]. Dystrophin deficiency is responsible for DAPC disintegration, with consequent widespread implications for muscle cell function. Another phenotype of dystrophin mutation is Becker muscular dystrophy (BMD), a relatively benign form of muscular dystrophy, with a semi-functional dystrophin [5].
The current clinical strategies for treating DMD are mainly symptomatic regimes, including glucocorticoids, physical therapy and symptomatic supportive therapy, which only delay the advancement of DMD, but do not halt or reverse its progression. In some conditions, these treatment options even have obvious side-effects [6]. Therefore, etiological therapeutic approaches for DMD have emerged; these therapeutic approaches either aim to directly restore dystrophin expression or indirectly target the downstream effectors caused by insufficient dystrophin [7]. As described below, the pathogenesis of DMD consists of multiple processes, thus therapies directly restoring dystrophin would resolve the original cause, while therapies targeting the downstream of insufficient dystrophin might only partially address these issues. This review focuses on reviewing the current development of the dystrophin gene-related nucleic acid therapies.
2. Pathogenic Mechanism of DMD
Since its first discovery in 1986, thousands of mutations of the dystrophin gene have been reported. Most patients with DMD carry one or more exon deletions; duplications, small deletions, insertions and point mutations are also frequently found, and these mutations all have their own “hot spot” regions [8,9]. These various mutations cause the production of non-functional dystrophin, which in turn accounts for the disassembly of DAPC. Because of the important role of DAPC in maintaining the structural integrity and contractile activity of muscle cells, its disintegration has a wide spectrum of adverse effects on muscle cell functions, such as sarcolemma weakening, functional ischemia, free-radical damage, cytosolic calcium overloading, regeneration failure, and sequences of muscle damage [10]. Multiple pathological processes are instigated and eventually the damaged muscle cells are replaced by fibrotic and adipose tissue, giving rise to muscle dysfunction.
3. Restoring Dystrophin Expression
The pathological changes of DMD are caused by the lack of functional dystrophin. Over the recent years, several therapies aimed at restoring dystrophin expression have been approved. The following novel nucleic acid therapeutic strategies have been developed to restore functional dystrophin expression (Figure 1).
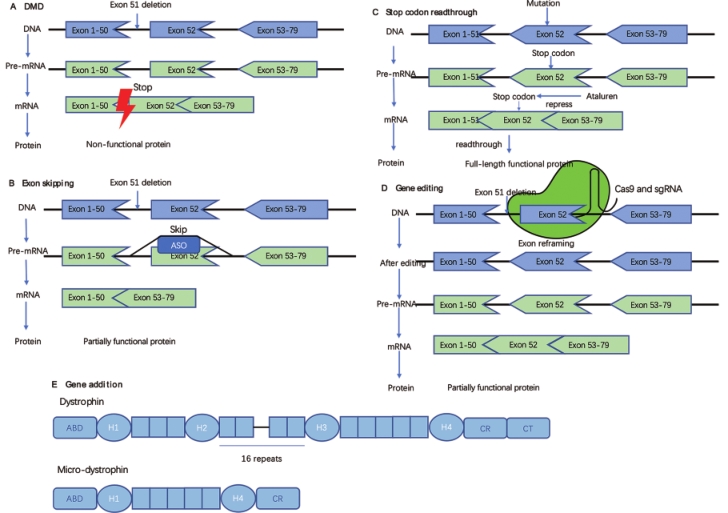
Figure 1. Approaches to restore dystrophin. A. Non-functional dystrophin generated by DMD. The deletion of exon 51 resulted in the production of a non-functional dystrophin. B. Exon skipping. A kind of antisense oligonucleotide (ASO) binds to exon 52 of the pre-mRNA of dystrophin, thus inducing skipping of exon 52 and restoring the reading frame. C. Stop codon readthrough. Ataluren can help patients with nonsense mutations readthrough the stop codon. D. Gene editing. One of the methods of gene editing: reframing the target exon to restore the reading frame. E. Gene addition. Comparison of the structure of a micro-dystrophin with that of a full-length dystrophin. ABD, actin-binding domain.
4. Exon Skipping
Antisense oligonucleotide (ASO) achieves exon skipping by regulating mRNA splicing, thereby restoring the disrupted reading frame and producing truncated, but functional dystrophin, similar to what happens in BMD. The first nucleic acid drug approved for DMD is Eteplisren, a phosphorodiamidate morpholino oligomer (PMO), that selectively binds to exon 51 of the pre-mRNA of dystrophin and re-instates the open reading frame. A phase III, multicenter, open-label study (NCT02255552) evaluated the efficacy and safety of Eteplirsen in a large cohort [11]. After 96 weeks, patients with DMD in the treatment group showed an increase in exon skipping (18.7-fold) and functional dystrophin (7-fold) compared to baseline. Consistent with the Phase II clinical trial (NCT01396239), the six-minute walk test distance decreased more slowly compared to the control group, and no drug-related adverse effects were observed [11,12]. In 2016, Eteplirsen was approved for marketing in the United States, even though the phase III clinical trial was not finished. Although there was incomplete evidence, advocation from patients contributed considerably to driving the drug development process [13].
The skipping efficiency of Eteplirsen could be improved by fusion with a cell-penetrating peptide [14]. Sarepta used this approach to develop SRP-5051; phase I trials have been completed and now it is in phase II for dose determination (NCT04004065). According to the information provided by Sarepta Therapeutics, SRP-5051 showed higher exon skipping efficiency and more dystrophin production at a tenfold lower exposure dose than Eteplirsen [15]. When SRP-5051 was administered at 30 mg/kg monthly, the mean exon skipping rate was 10.79% and the mean dystrophin expression was 6.55% [16]. However, safety is a prerequisite for drug trials; the U.S. Food and Drug Administration (FDA) placed SRP-5051 on clinical hold after two patients developed severe hypomagnesemia [17]. In September 2022, FDA removed the clinical hold on SRP-5051 and the trial will be conducted under more stringent safety monitoring [18].
In addition to Eteplirsen, there are three other ASO drugs already commercially available: Casimersen (exon 45), Viltolarsen (exon 53) and Golodirsen (exon 51). It is worth noting that the hot region of dystrophin gene mutations is exons 45–55, and if multi-exon skipping can be performed, more patients will benefit [19].
5. Stop Codon Read-Through
Approximately 13% of patients with DMD carry nonsense mutations with an early-appearing stop codon. Therefore, promoting read-through of the early-appearing stop codon might produce a fully functional protein. Ataluren is a small molecule with such a feature; it is able to bind to multiple protein synthesis apparatus sites and competitively inhibit release factor-dependent termination [20,21]. Data from a phase II trial (NCT00592553) concluded that Ataluren was statistically significantly better than placebo [22]. However, the outcomes of another phase II clinical trial (NCT03648827) showed minimal changes in muscle dystrophin levels in patients before and after the use of Ataluren. Moreover, the findings of a recent phase III clinical trial showed that the use of Ataluren delayed the age of ambulatory capacity loss by 2.2 years and the deterioration of predicted forced vital capacity to <60% by 3.0 years [23]. While results of a different phase III clinical trial (NCT01826487) showed that Ataluren only limited the loss of walking ability in some patients [24].
The efficacy of Ataluren remains controversial and its mechanism is not fully explored. Further studies are needed to identify suitable patients for this drug [25].
6. Gene Addition
Adeno-associated virus (AAV) vector has long been used when inserting exogenous genes for therapeutic purposes. However, the cDNA of the full-length dystrophin gene exceeds the packaging capacity of AAV, so a truncated micro-dystrophin or mini-dystrophin is used [26,27]. To minimize the toxicity and immunogenicity of expression in non-muscle tissues, a number of muscle-specific promoters have been tested for therapeutic use [28]. One of them, using the MHCK7 promoter (SRP-9001), has entered phase III clinical trial (NCT05096221). Four patients were recruited in the phase I/IIa and results showed no serious adverse effects after treatment. At 12 weeks post-trial, immunohistochemistry of gastrocnemius muscle biopsy specimens showed that a mean of 81.2% of myofibrils expressed micro-dystrophin, with a mean intensity of 96% at the sarcolemma. Western blot showed an average expression of 74.3% without fat or fibrosis adjustment [29]. In July 2022, Sarepta provided new data at the 17th International Congress on Neuromuscular Diseases (ICNMD 2022); the results of I/IIa trial (NCT03375164) showed that the total North Star Ambulatory Assessment (NSAA) score (the higher the score, the better the functional motor abilities, up to 34 points) was 9.9 points higher in patients treated over four years than that in the control group [30]. In another Open-Label trial (NCT04626674), after one year of treatment, the NSAA score was 3.8 points higher in the treatment group than that in the control group [30]. These data imply that patients in the treatment group have experienced a significant improvement in motor function. With positive clinical results, Sarepta announced in September 2022 that it has submitted a Biologics License Application (BAL) to FDA for an accelerated approval of SRP-9001.
Pfizer’s PF 06939926 (also in Phase III clinical trial) was suspended after one patient died in Phase I clinical trial [31]. Before this incident, a total of six serious adverse events were reported, including persistent vomiting resulting in dehydration, acute kidney injury with atypical hemolytic uremic syndrome (aHUS)-like complement activation, thrombocytopenia with aHUS-like complement activation [32] and myocarditis [33]. Evidence of active viral infection was found in these patients and after consultation with the study’s monitoring committee, the safety risk is deemed as “manageable” [34]. The trial resumed after Pfizer addressed the relevant requirements proposed by the FDA and updated the Phase III clinical trial protocol [35].
SGT-001, a gene therapy drug developed by Solid, was placed on hold twice due to reduced platelet and red blood cell counts in phase I/II trials. One patient also developed acute kidney injury, which could be a side effect caused by an overdose of the virus. The trial has resumed after all adverse events were resolved and once the safety management plan had been revised [36].
One concern is how long the micro-dystrophin transgene can be consistently expressed in humans after injection. In a canine model of DMD, gene expression and clinical score improvement were continuously maintained for up to 24 months after vector injection, in the absence of immunosuppression [37]. However, similar data is absent in patients with DMD. For patients with pre-existing AAV antibodies or those who have been treated but micro-dystrophin expression has declined, a change of vector serotype [38], plasma replacement or Imlifidase [39], which could reduce AAV antibody titer, is required.
In addition, common features of current micro-dystrophin coding sequences are the presence of an N-terminal domain, the cysteine-rich domain, a partial structure of the rod domain, and the absence of a C-terminal domain. The design of the sequence is related to the interaction of the expressed micro-dystrophin with various intracellular proteins, especially the rod domain. The rod domain may be associated with nNOS and possess the ability to regulate muscle perfusion and enhance muscle strength [40]. It also needs to be acknowledged that viral vectors are expensive and it is difficult to produce enough to meet patients’ need.
7. Gene Editing
The number of cells that need to be edited in patients with DMD is large, thus the ideal option is to deliver guide RNA and Cas9 to the appropriate location for in vivo editing. In vivo gene editing is suitable for DMD for several reasons: (1) the dystrophin gene is located on the X chromosome, so only one gene per nucleus needs to be corrected; (2) the dystrophin gene cannot be directly replaced due to its enormous size, but the gene itself can be repaired; (3) improving the patient’s condition does not require restoring dystrophin to normal level; and (4) the structure of the dystrophin gene allows restoration of some of its functions by exon skipping, which can also be achieved by gene editing [40]. With gene editing, exon remodeling, deletion and jumping can be performed. Base editing techniques can be used to regulate splicing or correct nonsense mutations, and even homology directed repair (HDR) can be performed [41]. One advantage of gene editing over gene addition is that the endogenous normal dystrophin can be regulated physically.
In multiple animal models of DMD, gene editing corrects mutations [40]. Recently, the first gene editing therapy, CRD-TMH-001, entered clinical trial (NCT05514249). It targets mutations in the dystrophin gene at the promoter or exon 1, which might stabilize or even reverse DMD progression by upregulating the expression of an isoform of dystrophin through gene editing [42].
At present, certain trials are ongoing for DMD (Table 1). ASO, stop codon read-through, gene addition and gene editing therapies all have considerable potential to help patients with DMD. However, there are many difficulties for nucleic acid therapies to overcome before they can be used clinically. The first challenge is safety; the immune response to the bacterial proteins and synthetic molecules used in gene editing must be minimized. The next challenge is effectiveness. Although the edited genes can be permanent, how long the cells will survive is still undefined. Exon skipping drugs require continual optimization of oligonucleotide sequences to increase skipping efficiency and reduce side effects. Finally, the delivery system might also trigger an immune response and side-effects [43]. Scientists and paralogical companies need to work in conjunction to address these challenges [44].
Table 1. Gene addition and gene editing therapies for DMD in ongoing clinical development.

8. Conclusion
Gene therapy has progressed rapidly since 1972, when Friedmann and Roblin proposed the hypothesis of gene therapy for the treatment of genetic diseases [45]. This has been especially important for rare diseases, where conventional drugs seldom prevent disease progression. Nucleic acid therapy based on genetic central dogma gives hope to patients. For the development and approval of drugs for rare diseases, it is important to consider clinical need, humanistic care and data-based scientific principles.
Author Contributions: G.H. drafted manuscript; G.H. prepared figures; C.C. supervised and drafted manuscript. All authors have read and agreed to the published version of the manuscript.
Funding: This work was supported by grants from the National Natural Science Foundation of China (82270363 to C.C.). No funding bodies had any role in the study design, data collection and analysis, decision to publish, or preparation of the article.
Data Availability Statement: Not applicable.
Acknowledgments: We thank our colleagues in Dr. Chen’s group for technical assistance and stimulating discussions during the course of this investigation.
Conflicts of Interest: The authors declare no conflict of interest.
References
- Crisafulli S.; Sultana J.; Fontana A.; et al. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet. J. Rare Dis., 2020, 15(1): 141. DOI: https://doi.org/10.1186/s13023-020-01430-8
- Mercuri E.; Bönnemann C.G.; Muntoni F. Muscular dystrophies. Lancet, 2019, 394(10213): 2025-38. DOI: https://doi.org/10.1016/S0140-6736(19)32910-1
- Zhao J.; Kodippili K.; Yue Y.; et al. Dystrophin contains multiple independent membrane-binding domains. Hum Mol Genet., 2016, 25(17): 3647-53. DOI: https://doi.org/10.1093/hmg/ddw210
- Gao Q.Q.; McNally E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol., 2015, 5(3): 1223-39. DOI: https://doi.org/10.1002/cphy.c140048
- Monaco A.P.; Bertelson CJ; Liechti-Gallati S; et.al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics, 1988, 2(1): 90-5. DOI: https://doi.org/10.1016/0888-7543(88)90113-9
- Gloss D.; Moxley R.T.; Ashwal S.; et.al. Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology, 2016, 86(5): 465-72. DOI: https://doi.org/10.1212/WNL.0000000000002337
- Verhaart I.E.C.; Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol.,2019, 15(7): 373-86. DOI: https://doi.org/10.1038/s41582-019-0203-3
- Aartsma-Rus A.; Van Deutekom J.C.T.; Fokkema I.F.; et.al. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve, 2006, 34(2): 135-44. DOI: https://doi.org/10.1002/mus.20586
- Bladen C.L.; Salgado D.; Monges S.; et al. The TREAT-NMD DMD Global Database: analysis of more than 7;000 Duchenne muscular dystrophy mutations. Hum. Mutat., 2015, 36(4): 395-402.
- Duan D.; Goemans N.; Takeda S.; et.al. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers., 2021, 7(1): 13. DOI: https://doi.org/10.1038/s41572-021-00248-3
- McDonald C.M.; Shieh P.B.; Abdel-Hamid H.Z.; et al. Open-Label Evaluation of Eteplirsen in Patients with Duchenne Muscular Dystrophy Amenable to Exon 51 Skipping: PROMOVI Trial. J. Neuromuscul. Dis., 2021, 8(6). DOI: https://doi.org/10.3233/JND-210643
- Mendell J.R.; Rodino-Klapac L.R.; Sahenk Z.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol., 2013, 74(5): 637-47. DOI: https://doi.org/10.1002/ana.23982
- Mattingly T.J.; Simoni-Wastila L. Patient-Centered Drug Approval: The Role of Patient Advocacy in the Drug Approval Process. J. Manag. Care Spec. Pharm., 2017, 23(10): 1078-82. DOI: https://doi.org/10.18553/jmcp.2017.23.10.1078
- Jirka S.M.G.; t Hoen P.A.C.; Diaz Parillas V.; et al. Cyclic Peptides to Improve Delivery and Exon Skipping of Antisense Oligonucleotides in a Mouse Model for Duchenne Muscular Dystrophy. Mol. Ther., 2018, 26(1): 132-47. DOI: https://doi.org/10.1016/j.ymthe.2017.10.004
- Sarepta Therapeutics I. Sarepta Therapeutics Announces Positive Clinical Results from MOMENTUM, a Phase 2 Clinical Trial of SRP-5051 in Patients with Duchenne Muscular Dystrophy Amenable to Skipping Exon 51. (accessed Oct 1,2022). https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-announces-positive-clinical-results.
- Sarepta Therapeutics I. Sarepta Therapeutics Reports Positive Clinical Results from Phase 2 MOMENTUM Study of SRP-5051 in Patients with Duchenne Muscular Dystrophy Amenable to Skipping Exon 51. (accessed Oct 1,2022). https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-reports-positive-clinical-results-phase-2.
- Sarepta Therapeutics I. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy. (accessed Oct 1,2022). https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-provides-update-srp-5051-treatment-duchenne.
- Sarepta Therapeutics I. Sarepta Therapeutics Announces That FDA has Lifted its Clinical Hold on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy. (accessed Oct 1,2022). https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-announces-fda-has-lifted-its-clinical-hold.
- Lee J.; Echigoya Y.; Duddy W.; et al. Antisense PMO cocktails effectively skip dystrophin exons 45-55 in myotubes transdifferentiated from DMD patient fibroblasts. PLoS One., 2018, 13(5): e0197084. DOI: https://doi.org/10.1371/journal.pone.0197084
- Ng M.Y.; Li H.; Ghelfi M.D., et al.. Ataluren and aminoglycosides stimulate read-through of nonsense codons by orthogonal mechanisms. Proc. Natl. Acad. Sci. USA, 2021, 118(2). DOI: https://doi.org/10.1073/pnas.2020599118
- Huang S.; Bhattacharya A.; Ghelfi M.D.; et al. Ataluren binds to multiple protein synthesis apparatus sites and competitively inhibits release factor-dependent termination. Nat. Commun., 2022, 13(1): 2413. DOI: https://doi.org/10.1038/s41467-022-30080-6
- Li D.; McDonald C.M.; Elfring G.L.; et al. Assessment of Treatment Effect With Multiple Outcomes in 2 Clinical Trials of Patients With Duchenne Muscular Dystrophy. JAMA Netw. Open, 2020, 3(2): e1921306. DOI: https://doi.org/10.1001/jamanetworkopen.2019.21306
- McDonald C.M.; Muntoni F.; Penematsa V.; et al. Ataluren delays loss of ambulation and respiratory decline in nonsense mutation Duchenne muscular dystrophy patients. J. Comp. Eff. Res., 2022, 11(3): 139-55. DOI: https://doi.org/10.2217/cer-2021-0196
- McDonald C.M.; Campbell C.; Torricelli R.E.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet, 2017, 390(10101): 1489-98. DOI: https://doi.org/10.1016/S0140-6736(17)31611-2
- Berger J.; Li M.; Berger S.; Effect of Ataluren on dystrophin mutations. J. Cell Mol Med., 2020, 24(12): 6680-9. DOI: https://doi.org/10.1111/jcmm.15319
- Wang B.; Li J.; Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. USA, 2000, 97(25): 13714-9. DOI: https://doi.org/10.1073/pnas.240335297
- Harper S.Q.; Hauser M.A.; DelloRusso C.; et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med., 2002, 8(3): 253-61. DOI: https://doi.org/10.1038/nm0302-253
- Duan D. Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther., 2018, 26(10): 2337-56. DOI: https://doi.org/10.1016/j.ymthe.2018.07.011
- Mendell J.R.; Sahenk Z;. Lehman K.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020, 77(9): 1122-31. DOI: https://doi.org/10.1001/jamaneurol.2020.1484
- Sarepta Therapeutics I. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies. (accessed Oct 1,2022). https://www.globenewswire.com/news-release/2022/07/06/2474964/36419/en/Sarepta-Therapeutics-Investigational-Gene-Therapy-SRP-9001-for-Duchenne-Muscular-Dystrophy-Demonstrates-Significant-Functional-Improvements-Across-Multiple-Studies.html.
- Pfizer I. Pfizer’s statement to the community. (accessed Oct 1,2022). https://www.parentprojectmd.org/wp-content/uploads/2021/12/DMD-Study-1001-Update_Letter-to-the-Community.pdf.
- Pfizer I. Pfizer’s New Phase 1b Results of Gene Therapy in Ambulatory Boys with Duchenne Muscular Dystrophy (DMD) Support Advancement into Pivotal Phase 3 Study. (accessed Oct 1,2022). https://investors.pfizer.com/Investors/News/news-details/2020/Pfizers-New-Phase-1b-Results-of-Gene-Therapy-in-Ambulatory-Boys-with-Duchenne-Muscular-Dystrophy-DMD-Support-Advancement-into-Pivotal-Phase-3-Study-05-15-2020/default.aspx.
- Pfizer I. A message from Pfizer on our DMD clinical program. (accessed Oct 1,2022). http://join.parentprojectmd.org/site/DocServer/A_Message_from_Pfizer_on_our_DMD_Clinical_Program_-_Sept.pdf.
- Pagliarulo N. Pfizer aims to restart late-stage trial of Duchenne gene therapy following safety setback. (accessed Oct 1,2022). https://www.biopharmadive.com/news/pfizer-duchenne-phase-3-study-reopen/618642/.
- Pfizer I. Pfizer announces clinical hold lifted for its mini-dystrophin gene therapy program. (accessed Oct 1,2022). https://www.parentprojectmd.org/wp-content/uploads/2022/04/Pfizer-Letter-to-the-Duchenne-community_April-28th-2022.pdf.
- Markati T.; Oskoui M.; Farrar M.A.; et al. Emerging therapies for Duchenne muscular dystrophy. Lancet Neurol. 2022, 21(9): 814-29. DOI: https://doi.org/10.1016/S1474-4422(22)00125-9
- Le Guiner C.; Servais L.; Montus M.; et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun., 2017, 8: 16105. DOI: https://doi.org/10.1038/ncomms16105
- Majowicz A.; Salas D.; Zabaleta N.; et al. Successful Repeated Hepatic Gene Delivery in Mice and Non-human Primates Achieved by Sequential Administration of AAV5 and AAV1. Mol. Ther., 2017, 25(8): 1831-42. DOI: https://doi.org/10.1016/j.ymthe.2017.05.003
- Leborgne C.; Barbon E.; Alexander J.M.; et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med., 2020, 26(7): 1096-101. DOI: https://doi.org/10.1038/s41591-020-0911-7
- Olson E.N. Toward the correction of muscular dystrophy by gene editing. Proc. Natl. Acad. Sci. USA, 2021, 118(22). DOI: https://doi.org/10.1073/pnas.2004840117
- Choi E.; Koo T. CRISPR technologies for the treatment of Duchenne muscular dystrophy. Mol. Ther., 2021, 29(11): 3179-91. DOI: https://doi.org/10.1016/j.ymthe.2021.04.002
- Johnson V. CRISPR Therapeutic Ready to Dose in Duchenne Muscular Dystrophy. (accessed Oct 1,2022). https://www.cgtlive.com/view/crispr-therapeutic-ready-to-dose-in-dmd.
- Kenjo E.; Hozumi H.; Makita Y.; et al. Low immunogenicity of LNP allows repeated administrations of CRISPR-Cas9 mRNA into skeletal muscle in mice. Nat. Commun., 2021, 12(1): 7101. DOI: https://doi.org/10.1038/s41467-021-26714-w
- Newby G.A.; Liu D.R. In vivo somatic cell base editing and prime editing. Mol. Ther., 2021, 29(11): 3107-24. DOI: https://doi.org/10.1016/j.ymthe.2021.09.002
- Friedmann T.; Roblin R. Gene therapy for human genetic disease? Science, 1972, 175(4025): 949-55. DOI: https://doi.org/10.1126/science.175.4025.949


