

Downloads
Download



This work is licensed under a Creative Commons Attribution 4.0 International License.
Review
Unveiling Catecholamine Dynamics in Cardiac Health and Disease: Mechanisms, Implications, and Future Perspectives
Wenjing Xiang, Xingyun Wang, Lei Li, Junhui Zeng, Haocheng Lu, and Ying Wang *
Department of Pharmacology, School of Medicine, Southern University of Science and Technology, Shenzhen 518055, China
* Correspondence: wangy6@sustech.edu.cn
Received: 15 August 2023
Accepted: 21 September 2023
Published: 27 December 2023
Abstract: Catecholamines play a pivotal role in regulating both cardiac physiology and pathology, orchestrating the “Fight-or-flight” response through the activation of sympathetic nervous system (SNS) activation and subsequent stimulation of adrenergic receptor. However, chronic stress and various cardiac diseases can disrupt catecholamine balance, contributing to cardiac dysfunction. The synthesis, release, reuptake, and degradation of catecholamines intricately regulate their concentration. Notably, catecholamine dynamics is markedly altered in heart diseases, including heart failure, myocardial infarction, and arrhythmias. While β-adrenergic receptor blockers, which block catecholamines from binding to the adrenergic receptors, are widely used in clinical settings, the potential implication of directly manipulating catecholamine homeostasis for the treatment of cardiac diseases have not been extensively explored. This review provides an overview of catecholaminergic systems, and discusses their intricate synthesis, release, uptake, and metabolism within the heart. Additionally, the review highlights mechanisms underlying cardiac effects of catecholamine dysregulation, including contractile dysfunction, electrical remodeling, and cardiac remodeling. Moreover, the review emphasizes the importance of considering spatiotemporal and sexual heterogeneity in catecholamine dynamics for cardiac precision medicine. In terms of future perspectives, we believe that harnessing genetically encoded fluorescent biosensors to map the heterogenous for real-time imaging of catecholamine dynamics and conducting gender-specific dissection of catecholamine dynamics have significant potential to advance personalized management of cardiac diseases management.
Keywords:
catecholamine dynamics cardiac diseases Förster resonance energy transfe (FRET)1. Introduction
Catecholamine dynamics play pivotal roles in both cardiac health and disease, constituting a paramount aspect of sympathetic regulation of the heart. Activation of adrenergic receptors (ARs) by catecholamines (norepinephrine, NE; epinephrine, EPI; and dopamine, DOPA) represents the primary mechanism for enhance cardiac performance in physiological conditions, whereas the sustained catecholamines stimulation is known as cardiotoxic [1‒3]. These physiological and pathological effects of catecholamines are mediated by α and β adrenergic receptors (ARs), with the βARs are thought to be the dominant myocytes ARs [4]. Therapeutics targeting to prevent the binding of endogenous catecholamines to βAR, known as β-blockers, have been widely used in the management of cardiac diseases, such as heart failure. Paradoxically, complicity of catecholamine dynamics and its implication in cardiac (patho-)physiology remain less understood, which hinders the development of pharmaceutical interventions directly targeting the modulation of catecholamine homeostasis. Notably, recent research advances indicate the significant heterogeneity of catecholamines dynamics underlying its fine-tuning of cardiac function in healthy and diseased conditions. Here, we will review the current knowledge and potential therapeutic implications of catecholamine dynamics, including its spatiotemporal and sexual heterogeneity, in the progression and management of cardiac diseases.
2. Catecholamine Dynamics: A Molecular Genetic Brief Overview of Catecholaminergic Systems
Catecholamine levels are balanced through precise regulation encompassing synthesis, release, uptake, and degradation [5]. This delicate equilibrium exerts a critical regulatory influence on both cardiac physiology and pathophysiology. Perturbations within the catecholamine pathway serve as a hallmark of distinct disease conditions, often involving specific molecular targets. The exploration of molecular mechanisms underlying catecholamine abnormalities holds significant promise for addressing cardiac diseases.
The biosynthesis of catecholamines commences with the conversion of the amino acid L-tyrosine into 3,4-dihydroxyphenylalanine (L-DOPA) through the action of tyrosine hydroxylase (TH) (Figure 1). Subsequently, aromatic L-amino acid decarboxylase converts L-DOPA to dopamine, stored within vesicles by the vesicular monoamine transporter [6]. The presence of dopamine β-hydroxylase (DβH) within storage vesicles leads to the conversion of dopamine to NE, preserving the appropriate noradrenergic phenotype of sympathetic nerves and CNS noradrenergic neurons. Norepinephrine N-methyltransferase (PNMT) catalyzes the N-methylation of NE to epinephrine (EPI) [7]. Upon release from nerve terminals, extracellular catecholamines undergo rapid clearance, primarily through uptake at these terminals. A small fraction (~5%) of catecholamines, including NE, can enter postsynaptic cells, such as cardiomyocytes, predominantly facilitated by corticosterone-sensitive OCT3 (organic cation transporter 3), also known as extraneuronal transporter monoamine transporter (EMT) [8,9]. OCT3 is necessary for translocating the catecholamines into cardiomyocytes and facilitate cardiac contractility. The majority of NE is cleared by neuronal uptake1 carrier protein, removing over 90% of NE from the cardiac synaptic cleft, which is facilitated by norepinephrine transporter (NET). A minor portion of NE is eliminated through extraneuronal carriers, uptake and circulatory dissipation [10]. NET, also known as the solute carriers 6 (SLC6) recycles catecholamines from synaptic spaces into presynaptic neurons while OCT3 is responsible for the peripheral elimination of catecholamines [11].

Figure 1. Catecholamine dynamics in the healthy and diseased heart. Production of catecholamine (norepinephrine, epinephrine, and dopamine) is initiated by tyrosine hydroxylase. Intracellular Calcium promotes the release of catecholamine from nerves into the synaptic cleft. Extracellular catecholamines bind to cardiac adrenergic receptors and triggers the downstream adrenergic signaling. Most of the extracellular catecholamines are quickly removed by the neurotransmitter transporter (NET)-mediated uptake1 and small portion of catecholamines enters the cardiomyocytes via extracellular monoamine transporter, including OCT3. Intracellular catecholamines are subjected to degradation by monoamine-oxidase A (MAO-A) and catechol-O-methyltransferase (COMT). These catecholamine processes are markedly changed in pathological conditions, including heart failure, myocardial infarction, and arrhythmias. The green arrow indicates the downregulation or upregulation of individual molecules in diseased conditions. Norepinephrine, NE; epinephrine, EPI; and dopamine, DOPA; 3,4-dihydroxyphenylalanine (L-DOPA).
Intracellularly, catecholamines undergo metabolism mediated by enzymes such as monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT). Two distinct MAO isoenzymes, MAOA and MAOB, metabolize NE and EPI into deaminated glycol metabolites, 3,4-dihydroxyphenylglycol (DHPG), and dopamine into 3,4-dihydroxyphenylacetic acid (DOPAC). COMT contributes by catalyzing O-methylation, converting NE to normetanephrine, EPI to metanephrine, and dopamine to 3-methoxytyramine (3-MT) [10]. This intricate catecholamine pathway is pivotal for maintaining physiological equilibrium and proper neurotransmitter function.
3. Catecholamines Dynamics Alteration in Heart Diseases
Catecholamine transmission undergoes significant alterations in various heart diseases, such as heart failure (HF), myocardial infraction (MI) and different forms of arrhythmias. These cardiac diseases are characterized by elevated plasma and interstitial catecholamines, with distinct mechanisms contributing to the dysregulation of catecholamines in each pathological condition.
3.1. Heart Failure
In congestive heart failure, the spillover of catecholamine into the plasma is notably amplified, exhibiting an eight-fold increase. Despite the elevated circulating catecholamine levels, the myocardial NE content is reduced in failing heart (Figure 1). This surge in circulating catecholamines can be primarily attributed to heightened sympathetic nerve activity and the resultant release of NE, coupled with compromised NE reuptake and uptake While the depletion of cardiac NE content arises from impaired catecholamine biosynthesis, uptake but elevated catabolism. Increased tyrosine hydroxylase (TH), the rate-limiting enzyme of catecholamine biosynthesis, is observed in HF. Similarly, in a HF rat model, reduction of NET binding sites impairs NE uptake and contributes to the reduced myocardial NE content [12]. Additionally, decreased NET densities and activities occur both in HF patients and animal models [13]. Overexpression of NET remarkably improve the structure and function of failing heart [14]. Elevated catecholamines metabolism is evident in both human and mouse failing hearts, with increased expression of cardiac MAOA and COMT established as key factors in the development of heart failure [15].
3.2. Myocardial Infarctions
MI is associated with multifaced alterations in catecholamines transmission. At the onset of a heart attack, ischemia triggers an acute stress response, leading to an increase in sympathetic nervous system activity. Consequently, there is an augmented release of catecholamines, particularly norepinephrine, from sympathetic nerve terminals in the heart. The ischemia injury also promotes sympathetic nerve remodeling in the heart, with the newly sprouting sympathetic fibers at ischemic area exhibit low NE content. Besides, inflammatory cytokines in MI heart aggravate the regional variation of myocardium NE levels by decreasing TH expression and thus leading to a local depletion of TH and impaired catecholamine biosynthesis in infarcted myocardium. Catecholamine metabolism is also altered in MI. Variation in the COMT gene have been associated with the risk of acute coronary events. After MI, elevation of MAO-A and COMT both indicates enhanced cytosolic catecholamine degradation [15,16].
3.3. Arrhythmias
Arrhythmogenesis involves both increased sympathetic drive and dysfunction within the heart [17‒19]. Increased concentration of circulating catecholamine contributes to the lethal ventricular arrythmias and subsequent sudden cardiac death. Rubart and Zipes found that the asynchronized heterogeneity of norepinephrine release within the heart contributes to arrythmia generation [20]. In experimental model, local application of NE induces premature ventricular complexes and ventricular tachycardia in intact hearts [21,22]. Defect or inhibition of the NET cause inadequate NE clearance and consequent excess sympathetic activation, resulting in a tachycardiac phenotype in humans. Furthermore, NET-/- mice display excessive tachycardia [23]. All the evidences indicate a critical NE disruption in arrhythmogenesis.
4. Mechanisms Underlying the Cardiovascular Effects of Catecholamines Dysregulation
Catecholamine dysregulation including the imbalance or abnormality in the catecholamine levels, process, or signaling contributes to various cardiac dysfunction through several mechanisms. Recent studies demonstrate that catecholamine-induced cardiotoxicity can be attributed to three major mechanisms: contractile dysfunction, electrical remodeling, and cardiac remodeling.
4.1. Contractile Dysfunction
Acute sympathetic activation enhances myocyte contraction while chronic catecholamine stimulation impairs contractility. It has been established that persistent catecholamine stimulation desensitizes the beta-adrenergic signaling by promoting receptor internalization and degradation, as well as inhibiting Gαs expression, thus disrupting myocyte contractility [24]. Interestingly, a recent study suggests myocyte cytosolic catecholamine (for instance NE) levels, which are tightly controlled by NE transport and degradation, are essential for cardiac contractile function [9]. A pool of intracellular β adrenergic receptors, newly identified located at the sarcoplasmic reticulum is activated by cytosolic rather than extracellular catecholamines. Disturbing catecholamine transport by inhibiting or deleting OCT3 dampens cardiac contractile response. Furthermore, elevated MAO-A reduces intracellular adrenergic receptor activation and impairs contractility. Inhibition or knockout of MAO-A enhances cardiac function [25]. The subcellular distribution of catecholamines and how it contributes to contractile dysfunction induced by sympathetic overactivation are interesting questions to be further investigated.
4.2. Electrical Remodeling
Electrical remodeling constitutes a pivotal mechanism through which catecholamines promote various arrhythmias, such as atrial fibrillation and ventricular arrhythmia. The electrical remodeling driven by catecholamines consists of arrhythmogenic calcium overload, ion channel irregulation and the disruption of synchronization. The excessive activation of adrenergic receptor-coupled G-proteins, along with downstream cAMP-PKA signaling, fosters an increased influx of calcium into the calcium channel and triggers calcium release from RyR receptors. This, in turn, leads to a state of calcium overload, facilitating the emergence of arrhythmogenic calcium waves and cascading sparks [26]. Furthermore, excessive catecholamines disrupts channel expression and suppresses the repolarizing potassium (K+) current and alters myocyte action potential [18,27]. The disruption of local sympathetic transmission within the heart also contributes to the development of arrhythmias [28]. Clinically, the deficiency of the NET in the context of MI has been linked to severe ventricular arrhythmias [29,30]. Moreover, an elevated sensitivity to NE and increased dispersion of repolarization, driven by sympathetic denervation in proximity to the scar tissue, play a significant role in arrhythmogenesis. Electrical remodeling of myocytes, influenced by inappropriate heterogeneity in NE release, culminates in cardiac electric instability and significantly contributes to the occurrence of arrhythmias.
4.3. Cardiac Remodeling
Excessive catecholamine signaling is widely recognized as a driver of cardiac remodeling, involving mechanisms such as increased oxygen demand, heightened inflammation, fibrosis, and the generation of reactive oxygen species (ROS). An overactive sympathetic drive triggers the release of catecholamines, intensifying the cardiac workload by elevating heart rate and contractility [3]. Consequently, this disruption in the delicate balance between oxygen supply and demand within the myocardium culminates in cardiac hypertrophy and dilation [31‒33]. These pathological alterations significantly contribute to the progression of HF and hypertensive heart diseases. Experimental administration of high doses of NE induces cardiac remodeling, effectively mirroring the effects of chronic catecholamine stimulation in humans. Intravenous infusion of NE to rodents induces hypertrophy in rats and is associated with elevated levels of proinflammatory cytokines and fibrosis [34,35]. In rat hearts subjected to NE infusion, there is an observable elevation in interleukin-1β, interleukin-6 mRNA, as well as fibrotic collagens and natriuretic peptide [35,36]. Interestingly, this induced fibrosis is specific to the left ventricle and not observed in the right ventricle. While it is established that excessive catecholamine signaling promotes cardiac remodeling, the complete mechanistic underpinning remains to be fully elucidated. Among the most extensively studied mechanisms of catecholamine-dependent cardiomyopathy is the generation of ROS [19,37,38]. The oxidation of catecholamines at the mitochondria by MAO gives rise to ROS [39,40]. In instances of MI and failing hearts, MAO-A is considered a major source of cardiac oxidative stress and undergoes notable upregulation [25]. The reduction of ROS through MAOA inhibition or the use of vitamin C demonstrates protective effects against stress-induced heart damage [41]. Elevated catecholamine oxidation and degradation result in the excessive production of oxidative stress. Both calcium overload and ROS have the potential to instigate cardiomyocyte death through mitochondrial dysfunction. Intriguingly, research advances suggest that, the nuclear adrenergic signaling promotes hypertrophic transcriptional factor activity including ERK and SMAD, demonstrating that catecholamine stimulation promote cardiac hypertrophy through a local adrenergic signaling microdomain [42,43]. How the enzymes for catecholamine synthesis, release, uptake and metabolism could affect this subcellular microdomain remain to be elucidated.
5. Precision Medicine Targeting Catecholamines Dynamics
The heterogeneity of catecholamines dynamics exists in various aspects, including spatial, temporal, and gender-related variations. Analyzing and understanding this heterogeneity is crucial for achieving precise regulation of catecholamine dynamics, and it has been largely overlooked for a long time in previous cardiac studies. Recent findings have unraveled novel roles and molecular mechanisms governing the regulation of catecholamines regulation within the heart. Catecholamine processing is a dynamic with spatiotemporal specificity within the heart. At the tissue level, regional concentrations, release patterns, catecholamine receptors and signaling regulation across the heart are not uniform. For instance, both the catecholamines concentrations and distribution of catecholamine receptors are uneven in the heart. αARs are heterogeneously distributed within the heart, with mRNA transcription level of αAR displays the following regional rank order: left ventricle > left atrium > apex > right atria [44]. Pathological conditions, such as heart failure, can alter the spatial distribution of adrenergic receptors in human hearts [45]. Within a single cell, catecholamine biogenesis, release, metabolism, and uptake occur at specialized subcellular compartment. As a result, there is a gradient of catecholamines levels across different compartments (plasma membrane, cytoplasm, mitochondrial outer membrane, etc.). Local catecholamines exert diverse physiological and pathological effects by activating subcellular-localized catecholamine receptors, which are emerging as a new area for future investigation of catecholamine systems. However, these regional differences are often not taken into account in numerous studies of catecholamine-induced cardiac contraction or cardiac injury, spanning from the organ level (the heart) down to the subcellular level.
Cardiac disease often exhibits sex-specific patterns, yet sexual heterogeneity has been inadequately explored in previous cardiac studies. Notably, females and males can experience differences of catecholamine concentrations, gene transcription and the effects of catecholamines on cardiac contraction and heart rhythm. Understanding how catecholamines impacts cardiac function differently in men and women is required for developing personalized and effective therapeutic interventions [46,47].
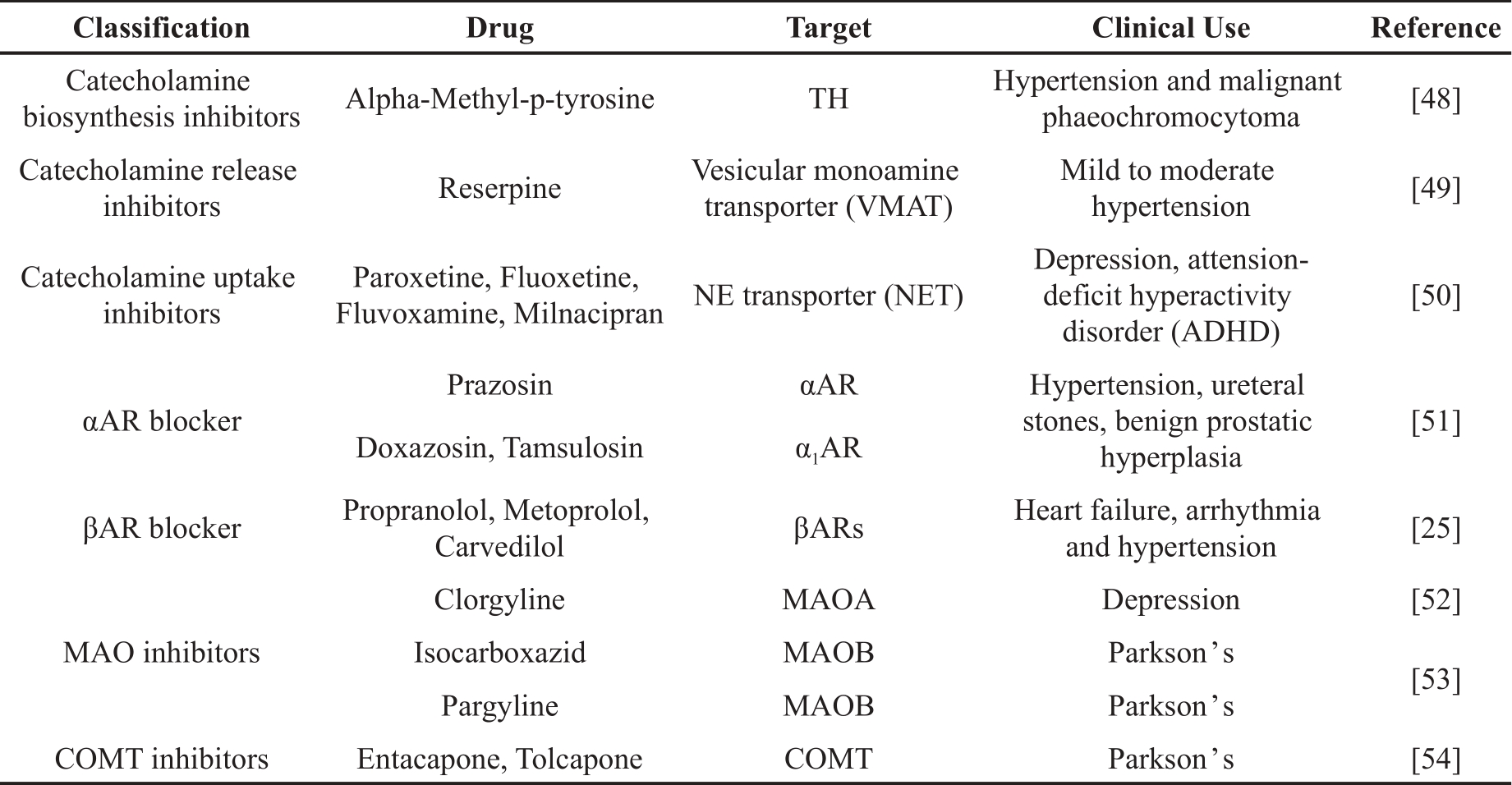
A substantial number of drugs that influences catecholamine dynamics are currently employed in clinical treatment of neuronal disorders and vascular dysfunction. In this context, we provide a brief summary of various subgroup of drugs intervening catecholamine biosynthesizing, releasing, uptake, metabolism and its binding to adrenergic receptors (Table 1) [25,48‒54]. Given the spatiotemporal and sexual heterogeneity observed in catecholamines dynamics, it is important to note that these drugs may also exhibit sex-specific or subcellular-specific effects, necessitating further investigation in the future research.
Table 1. Drugs targeting catecholamine dynamics.
6. Future Directions
6.1. Spatiotemporal Dynamics of Catecholamines Illuminated by Genetically Encoded Fluorescent Biosensor
Within cells and tissues, it is important to note that catecholamines are not uniformly distributed and undergo rapid processing and degrading. Spatiotemporal alteration of catecholamines is closely linked to a range of diseases, including neuronal disorders and cardiac diseases. Nevertheless, the real-time monitoring of the spatiotemporal dynamics catecholamines in subcellular resolution poses a significant challenge. This challenge underscores the need for a comprehensive understanding of how catecholamine dynamics influences both physiological and pathological processes and for the development of therapeutic interventions aimed at manipulating catecholamines at subcellular level. Indeed, the dynamics of catecholamines in neurons and brain have been unraveled with the development of catecholamine sensors [55]. In contrast, less is known about the dynamics in cardiomyocytes and within the heart. In this section, we provide a comprehensive synthesis of recent advancements and offer insights into prospects pertaining to utilizing genetically encoded sensors to facilitate real-time imaging of catecholamine dynamics within the heart.
Over the past two decades, a diverse array of sensors has been devised to the capture rapid fluctuations in catecholamine levels with exceptional spatial and temporal precision [56]. These biosensors are broadly categorized into two main groups: those that directly detect catecholamine binding and those that monitor downstream signaling pathways activated by catecholamines. One prominent approach employs fluorescent resonance energy transfer (FRET)-based biosensors that discern catecholamines through the detection of conformational alterations induced upon binding to G protein-coupled receptors (GPCRs) [57]. By incorporating a pair of FRET proteins into the intracellular loop and C-terminal domain of the β2-adrenergic receptor, these biosensors facilitate millisecond-scale monitoring of NE concentration changes. Besides, a novel genetically encoded GPCR-activation-based norepinephrine sensors (GRABNE) by converting the conformation change between the fifth and sixth transmembrane domains in response to NE binding to modulate fluorescence protein brightness. This new sensor (GRABNE) has enabled highly specific in vivo measurement with high spatiotemporal resolution of NE dynamics in zebrafish and mice brain [58].
In contrast to the direct assessment of conformational shifts triggered by catecholamine-receptor binding, an alternative approach involves engineered fluorescent reporters that gauge downstream signaling molecules propagated by adrenergic receptors. Specifically, a suite of cell-based catecholamine neurotransmitter fluorescent engineered receptors (CNiFERs) has been developed to monitor intracellular calcium fluctuations following neurotransmitter interactions [59]. Moreover, a range of cAMP and PKA biosensors have been strategically deployed to trace regional adrenergic receptor activation-transduced cAMP-PKA signaling in response to catecholamine stimulation [2,60]. The use of subcellular-localized biosensors has unveiled heterogeneous activation of adrenergic receptors by catecholamines such as NE and EPI across distinct cellular compartments [8,9]. Additionally, the intriguing discovery that cytosolic catecholamines transported by OCT3 activate intracellular receptors differently from conventional cell surface receptors underscores the complexity of catecholamine effects within the heart [9]. Furthermore, to convert catecholamine-induced receptor activation into a sustained intracellular signal, the TANGO assay has been developed [61]. This innovative approach involves fusing the transcription factor, tetracycline-controlled transactivator (TA), to the receptor's C-terminal, thereby initiating reporter gene expression. The TANGO assay boasts single-cell resolution and demonstrates nanomolar sensitivity upon neurotransmitter-induced activation (Figure 2).
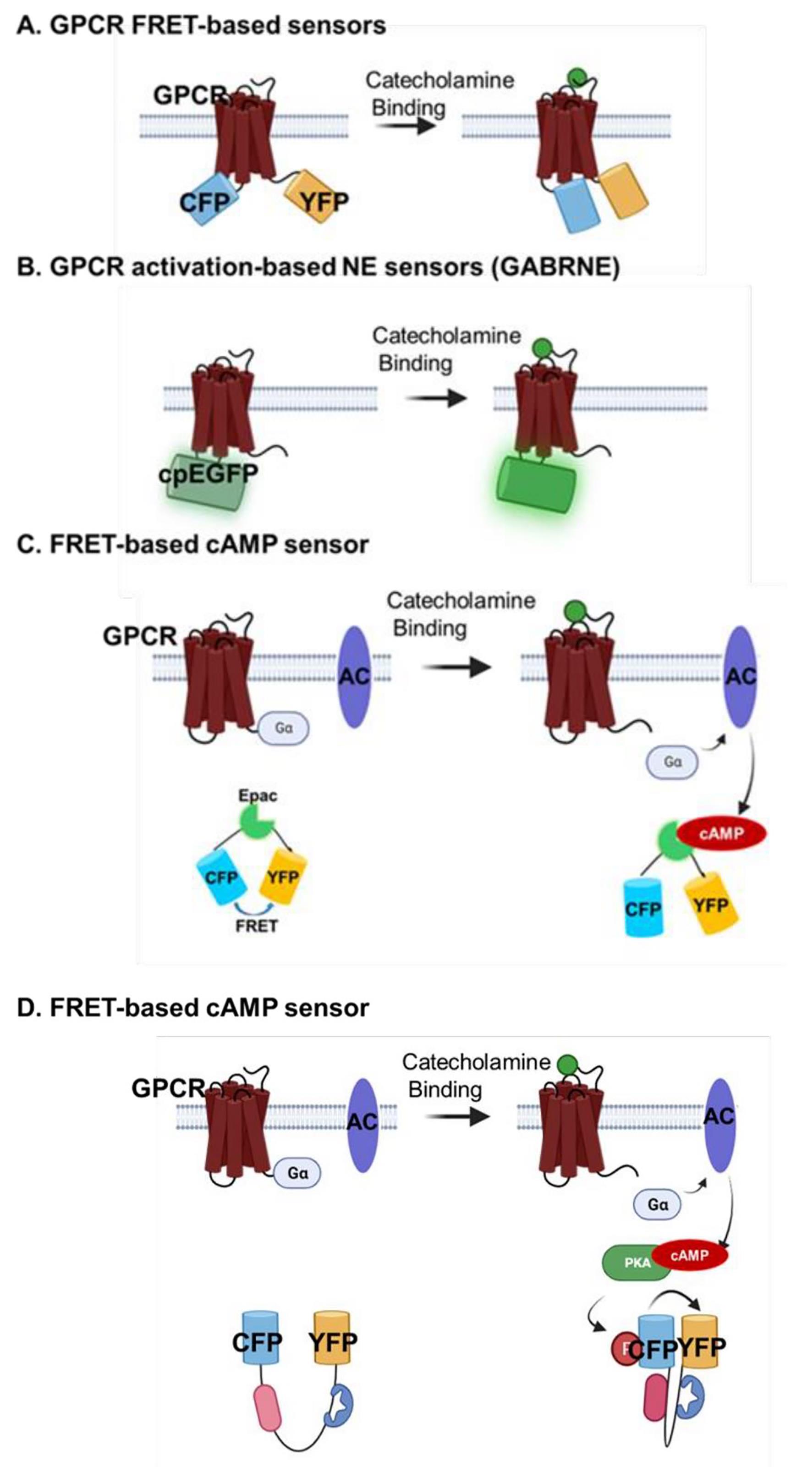

Figure 2 Biosensors for illuminating catecholamines signals. The design and working principle showing how distinct sensor detect catecholamines or it-activated signaling pathway. A. GPCR FRET-based biosensors. B. GPCR activation-based NE sensors C. FRET-based D. FRET-based PKA; E TANGO assay. Fluorescence resonance energy transfer: FRET; Norepinephrine: NE; Protein kinase A: PKA; Transcriptional factor: TA.
To date, various pioneering research tools for monitoring the dynamics of catecholamines transmission has been developed and applied to central neuron system in vivo [58] and in vitro. However, these tools have less deployed to unravel the complexities of catecholamine dynamics within the heart at whole-heart level. Further exploration of these techniques in cardiac contexts holds significant promise for expanding our understanding of catecholamine dynamics and their impact in heart function.
6.2. Sex Differences
The prevalence, presentation and drug-responses of various cardiac diseases exists between males and females. Understanding the mechanisms underlying these differences is essential for customized precision treatment [62‒64]. Interestingly, Sexual dimorphism in the distribution, function, and regulation of catecholamines is a complex phenomenon that encompasses various aspects of neurotransmitter activity and metabolism [65‒67]. This interplay between sex and catecholamines has been elucidated through a range of studies, which concerns distinct patterns of release, enzymatic activity, and receptor response between males and females. Moreover, sex dimorphism in the function, regulation and distribution of catecholamines is a complex phenomenon that encompasses various aspects of neurotransmitter activity and metabolism. The implications of this sex dimorphism in the realm of cardiac diseases remain an area warranting further exploration.
The landscape of sex-related catecholamine dynamics is multifaceted, as evidenced by pronounced sex-specific disparities in catecholamine release through exocytosis, revealed by single-vesicle electrochemistry [65]. Notably, male rats have exhibited higher catecholamine release rates and an increased frequency of exocytotic events. This phenomenon is paralleled in clinical observations, as male hypertensive patients display augmented sympathetic nervous system (SNS) activity and elevated urine catecholamine levels, comparing to their female counterparts. These revelations underline the necessity for tailoring hypertensive treatment strategies to account for sex-dependent variations. As evidenced by the distinctive sympathetic activation observed in women patients with and without HF [66]. Notably, coronary sinus plasma analysis has demonstrated elevated NE concentrations in women, emphasizing cardiac-specific sympathetic engagement. Furthermore, gender-dependent catecholamine metabolism emerges as an additional contributor to the intricate tapestry of sex dimorphism within catecholamine systems. Enzymes responsible for catecholamine metabolism exhibit conspicuous sexual dimorphism, a fact underscored by the sexual dimorphism of MAO-A and its X-chromosomal location [68]. This enzymatic variance extends to COMT, whose activity is attenuated in women due to estrogen-mediated inhibition of COMT mRNA expression [69].
It is noteworthy that the role and potential therapeutic target concerning the sex-dependent catecholamines transmission in cardiac diseases, including HF, MI and arrhythmias represent as a frontier awaiting comprehensive investigation.
7. Conclusions
Catecholamine dynamics plays a crucial role in various physiological and pathological processes of the heart. Investigating mechanisms and potential therapeutic avenues, including sex-specific considerations and innovative biosensing techniques, holds promise for advancing cardiac precision medicine.
Author Contributions: Conceptualization, Y. W. and H.L.; software, Y.W.; investigation, W.X. and J.Z.; writing—original draft preparation, W.X., L.L. and Y.W.; writing—review and editing, W.X., X.W. and Y.W.; visualization, W.X., L.L and X.W.; supervision, Y.W.; project administration, Y.W. All authors have read and agreed to the published version of the manuscript.
Funding: This research received no external funding.
Data Availability Statement: Not applicable.
Acknowledgments: We acknowledge Haibo Ni for his generous contributions in language editing and intellectual refinement of this manuscript.
Conflicts of Interest: The authors declare no conflict of interest.
References
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. DOI: https://doi.org/10.1038/415198a
- Barbagallo, F.; Xu, B.; Reddy, G.R.; et al. Genetically Encoded Biosensors Reveal PKA Hyperphosphorylation on the Myofilaments in Rabbit Heart Failure. Circ. Res. 2016, 119, 931–943. DOI: https://doi.org/10.1161/CIRCRESAHA.116.308964
- Liaudet, L.; Calderari, B.; Pacher, P. Pathophysiological mechanisms of catecholamine and cocaine-mediated cardiotoxicity. Heart Fail. Rev. 2014, 19, 815–824. DOI: https://doi.org/10.1007/s10741-014-9418-y
- Song, Y.; WOO, AY-H.; Zhang, Y.; et al. Cardiac β-Adrenoceptor signaling: The new insight on an old target in the therapy of cardiovascular disease. IJDDP 2022, 1, 3. DOI: https://doi.org/10.53941/ijddp.v1i1.177
- Kvetnansky, R.; Sabban, E.L.; Palkovits, M. Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol, Rev. 2009, 89, 535–606. DOI: https://doi.org/10.1152/physrev.00042.2006
- Ebert, S.N.; Rong, Q.; Boe, S.; et al. Catecholamine-synthesizing cells in the embryonic mouse heart. Ann, N, Y, Acad, Sci. 2008, 1148, 317–324. DOI: https://doi.org/10.1196/annals.1410.008
- Tillinger, A.; Novakova, M.; Pavlovicova, M.; et al. Modulation by 6-hydroxydopamine of expression of the phenylethanolamine N-methyltransferase (PNMT) gene in the rat heart during immobilization stress. Stress 2006, 9, 207–213. DOI: https://doi.org/10.1080/10253890601069385
- Nash, C.A.; Wei, W.; Irannejad, R.; et al. Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. Elife 2019, 8, e48167. DOI: https://doi.org/10.7554/eLife.48167
- Wang, Y.; Shi, Q.; Li, M.; et al. Intracellular β1-Adrenergic Receptors and Organic Cation Transporter 3 Mediate Phospholamban Phosphorylation to Enhance Cardiac Contractility. Circ. Res. 2021, 128, 246–261. DOI: https://doi.org/10.1161/CIRCRESAHA.120.317452
- Eisenhofer, G.; Kopin, I.J.; Goldstein, D.S. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol. Rev. 2004, 56, 331–349. DOI: https://doi.org/10.1124/pr.56.3.1
- Schlessinger, A.; Geier, E.; Fan, H.; et al. Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 15810–15815. DOI: https://doi.org/10.1073/pnas.1106030108
- Backs, J.; Haunstetter, A.; Gerber, S.H.; et al. The neuronal norepinephrine transporter in experimental heart failure: evidence for a posttranscriptional downregulation. J. Mol. Cell. Cardiol. 2001, 33, 461–472. DOI: https://doi.org/10.1006/jmcc.2000.1319
- Münch, G.; Rosport, K.; Bültmann, A.; et al. Cardiac overexpression of the norepinephrine transporter uptake-1 results in marked improvement of heart failure. Circ. Res. 2005, 97, 928–936. DOI: https://doi.org/10.1161/01.RES.0000186685.46829.E5
- Recchia, F.A.; Giacca, M. Targeted uptake-1 carrier to rescue the failing heart. Circ. Res. 2005, 97, 847–849. DOI: https://doi.org/10.1161/01.RES.0000190404.47996.7d
- Wang, Y.; Zhao, M.; Shi, Q.; et al. Monoamine Oxidases Desensitize Intracellular β1AR Signaling in Heart Failure. Circ. Res. 2021, 129, 965–967. DOI: https://doi.org/10.1161/CIRCRESAHA.121.319546
- Hall, K.T.; Battinelli, E.; Chasman, D.I. Catechol-O-Methyltransferase and Cardiovascular Disease: MESA. J. Am. Heart Assoc. 2019, 8, e014986. DOI: https://doi.org/10.1161/JAHA.119.014986
- Motiejunaite, J.; Amar, L.; Vidal-Petiot, E. Adrenergic receptors and cardiovascular effects of catecholamines. Ann. Endocrinol (Paris). 2021, 82, 193–197. DOI: https://doi.org/10.1016/j.ando.2020.03.012
- Gardner, R.T.; Ripplinger, C.M.; Myles, R.C.; et al. Molecular Mechanisms of Sympathetic Remodeling and Arrhythmias. Circ. Arrhythm. Electrophysiol. 2016, 9, e001359. DOI: https://doi.org/10.1161/CIRCEP.115.001359
- Francis Stuart, S.D.; Wang, L.; Woodard, W.R.; et al. Age-related changes in cardiac electrophysiology and calcium handling in response to sympathetic nerve stimulation. J. Physiol. 2018, 596, 3977–3991. DOI: https://doi.org/10.1113/JP276396
- Rubart, M.; Zipes, D.P. Mechanisms of sudden cardiac death. J. Clin. Invest. 2005, 115, 2305–2315. DOI: https://doi.org/10.1172/JCI26381
- Myles, R.C.; Wang, L.; Kang, C.; et al. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ. Res. 2012, 110, 1454–1464. DOI: https://doi.org/10.1161/CIRCRESAHA.111.262345
- Myles, R.C.; Wang, L.; Bers, D.M.; et al. Decreased inward rectifying K+ current and increased ryanodine receptor sensitivity synergistically contribute to sustained focal arrhythmia in the intact rabbit heart. J. Physiol. 2015, 593, 1479–1493. DOI: https://doi.org/10.1113/jphysiol.2014.279638
- Keller, N.R.; Diedrich, A.; Appalsamy, M.; et al. Norepinephrine transporter-deficient mice exhibit excessive tachycardia and elevated blood pressure with wakefulness and activity. Circulation 2004, 110, 1191–1196. DOI: https://doi.org/10.1161/01.CIR.0000141804.90845.E6
- Xiao, R.P.; Zhang, S.J.; Chakir, K.; et al. Enhanced G(i) signaling selectively negates beta2-adrenergic receptor (AR)--but not beta1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation 2003, 108, 1633–1639. DOI: https://doi.org/10.1161/01.CIR.0000087595.17277.73
- Wang, Y.; Zhao, M.; Xu, B.; et al. Monoamine oxidase A and organic cation transporter 3 coordinate intracellular β1AR signaling to calibrate cardiac contractile function. Basic Res. Cardiol. 2022, 117, 37. DOI: https://doi.org/10.1007/s00395-022-00944-5
- Xu, B.; Li, M.; Wang, Y.; et al. GRK5 Controls SAP97-Dependent Cardiotoxic β1 Adrenergic Receptor-CaMKII Signaling in Heart Failure. Circ. Res. 2020, 127, 796–810. DOI: https://doi.org/10.1161/CIRCRESAHA.119.316319
- Pogwizd, S.M.; Schlotthauer, K.; Li, L.; et al. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ. Res. 2001, 88, 1159–1167. DOI: https://doi.org/10.1161/hh1101.091193
- Qin, F.; Vulapalli, R.S.; Stevens, S.Y.; et al. Loss of cardiac sympathetic neurotransmitters in heart failure and NE infusion is associated with reduced NGF. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H363–H371. DOI: https://doi.org/10.1152/ajpheart.00319.2001
- Stanton, M.S.; Tuli, M.M.; Radtke, N.L.; et al. Regional sympathetic denervation after myocardial infarction in humans detected noninvasively using I-123-metaiodobenzylguanidine. J. Am. Coll. Cardiol. 1989, 14, 1519–1526. DOI: https://doi.org/10.1016/0735-1097(89)90391-4
- Hartikainen, J.; Kuikka, J.; Mäntysaari, M.; et al. Sympathetic reinnervation after acute myocardial infarction. Am. J. Cardiol. 1996, 77, 5–9. DOI: https://doi.org/10.1016/S0002-9149(97)89125-4
- Eschenhagen, T. Is Stimulation of Cardiomyocyte Renewal a Facette of Reversible Catecholamine Toxicity? Circ. Res. 2016, 119, 779–781. DOI: https://doi.org/10.1161/CIRCRESAHA.116.309663
- Wallner, M.; Duran, J.M.; Mohsin, S.; et al. Acute Catecholamine Exposure Causes Reversible Myocyte Injury Without Cardiac Regeneration. Circ. Res. 2016, 119, 865–879. DOI: https://doi.org/10.1161/CIRCRESAHA.116.308687
- Rona, G. Catecholamine cardiotoxicity. J. Mol. Cell. Cardiol. 1985, 17, 291–306. DOI: https://doi.org/10.1016/S0022-2828(85)80130-9
- Du, Y.; Demillard, L.J.; Ren, J. Catecholamine-induced cardiotoxicity: A critical element in the pathophysiology of stroke-induced heart injury. Life Sci. 2021, 287, 120106. DOI: https://doi.org/10.1016/j.lfs.2021.120106
- Xiao, H.; Li, H.; Wang, J.J.; et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur. Heart, J. 2018, 39, 60–69. DOI: https://doi.org/10.1093/eurheartj/ehx261
- Meier, H.; Bullinger, J.; Marx, G.; et al. Crucial role of interleukin-6 in the development of norepinephrine-induced left ventricular remodeling in mice. Cell. Physiol. Biochem. 2009, 23, 327–334. DOI: https://doi.org/10.1159/000218180
- Wang, Y.; Hu, H.; Yin, J.; et al. TLR4 participates in sympathetic hyperactivity Post-MI in the PVN by regulating NF-κB pathway and ROS production. Redox. Biol. 2019, 24, 101186. DOI: https://doi.org/10.1016/j.redox.2019.101186
- Costa, V.M.; Carvalho, F.; Bastos, M.L.; et al. Contribution of catecholamine reactive intermediates and oxidative stress to the pathologic features of heart diseases. Curr. Med. Chem. 2011, 18, 2272–2314. DOI: https://doi.org/10.2174/092986711795656081
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; et al. Parini A, Di Lisa, F. Monoamine oxidases as sources of oxidants in the heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. DOI: https://doi.org/10.1016/j.yjmcc.2013.12.032
- Di Sante, M.; Antonucci, S.; Pontarollo, L.; et al. Monoamine oxidase A-dependent ROS formation modulates human cardiomyocyte differentiation through AKT and WNT activation. Basic Res. Cardiol. 2023, 118, 4. DOI: https://doi.org/10.1007/s00395-023-00977-4
- Kaludercic, N.; Carpi, A.; Menabò R.; et al. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta. 2011, 1813, 1323–1332. DOI: https://doi.org/10.1016/j.bbamcr.2010.09.010
- Wright, C.D.; Chen, Q.; Baye, N.L.; et al. Nuclear alpha1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ. Res. 2008, 103, 992–1000. DOI: https://doi.org/10.1161/CIRCRESAHA.108.176024
- Subramaniam, G.; Schleicher, K.; Kovanich, D.; et al. Integrated Proteomics Unveils Nuclear PDE3A2 as a Regulator of Cardiac Myocyte Hypertrophy. Circ. Res. 2023, 132, 828–848. DOI: https://doi.org/10.1161/CIRCRESAHA.122.321448
- Wolff, D.W.; Dang, H.K.; Liu, M.F.; et al. Distribution of alpha1-adrenergic receptor mRNA species in rat heart. J. Cardiovasc. Pharmacol. 1998, 32, 117–122. DOI: https://doi.org/10.1097/00005344-199807000-00018
- Brodde, O.E.; Bruck, H.; Leineweber, K.; et al. Presence, distribution and physiological function of adrenergic and muscarinic receptor subtypes in the human heart. Basic Res. Cardiol. 2001, 96, 528–538. DOI: https://doi.org/10.1007/s003950170003
- Ostadal, B.; Ostadal, P. Sex-based differences in cardiac ischaemic injury and protection: therapeutic implications. Br. J. Pharmacol. 2014, 171, 541–554. DOI: https://doi.org/10.1111/bph.12270
- Ji, H.; Kwan, A.C.; Chen, M.T.; et al. Sex Differences in Myocardial and Vascular Aging. Circ. Res. 2022, 130, 566–577. DOI: https://doi.org/10.1161/CIRCRESAHA.121.319902
- Brogden, R.N.; Heel, R.C.; Speight, T.M.; et al. alpha-Methyl-p-tyrosine: a review of its pharmacology and clinical use. Drugs, 1981, 21, 81–89. DOI: https://doi.org/10.2165/00003495-198121020-00001
- Mandela, P.; Chandley, M.; Xu, Y.Y.; et al. Reserpine-induced reduction in norepinephrine transporter function requires catecholamine storage vesicles. Neurochem. Int. 2010, 56, 760–767. DOI: https://doi.org/10.1016/j.neuint.2010.02.011
- Bourin, M.; Chue, P.; Guillon, Y. Paroxetine: a review. CNS Drug Rev. 2001, 7, 25–47. DOI: https://doi.org/10.1111/j.1527-3458.2001.tb00189.x
- Guo, J.; Tang, R. Efficacy and tolerability of doxazosin gastro-intestinal therapeutic system versus tamsulosin in patients with lower urinary tract symptoms associated with benign prostatic hyperplasia: A systematic review and meta-analysis. Medicine (Baltimore) 2021, 100, e26955. DOI: https://doi.org/10.1097/MD.0000000000026955
- Wang, Q.; Wang, Y.; West, T.M.; et al. Carvedilol induces biased β1 adrenergic receptor-nitric oxide synthase 3-cyclic guanylyl monophosphate signalling to promote cardiac contractility. Cardiovasc. Res. 2021, 117, 2237–2251. DOI: https://doi.org/10.1093/cvr/cvaa266
- Guillem, K.; Vouillac, C.; Azar, M.R.; et al. Monoamine oxidase inhibition dramatically increases the motivation to self-administer nicotine in rats. J. Neurosci. 2005, 25, 8593–8600. DOI: https://doi.org/10.1523/JNEUROSCI.2139-05.2005
- Graf, W.D.; Unis, A.S.; Yates, C.M.; et al. Catecholamines in patients with 22q11.2 deletion syndrome and the low-activity COMT polymorphism. Neurology 2001, 57, 410–416. DOI: https://doi.org/10.1212/WNL.57.3.410
- Jing, M.; Zhang, Y.; Wang, H.; et al. G-protein-coupled receptor-based sensors for imaging neurochemicals with high sensitivity and specificity. J. Neurochem. 2019, 151, 279–288. DOI: https://doi.org/10.1111/jnc.14855
- Wang, H.; Jing, M.; Li, Y. Lighting up the brain: genetically encoded fluorescent sensors for imaging neurotransmitters and neuromodulators. Curr. Opin. Neurobiol. 2018, 50, 171–178. DOI: https://doi.org/10.1016/j.conb.2018.03.010
- Nakanishi, J.; Takarada, T.; Yunoki, S.; et al. FRET-based monitoring of conformational change of the beta2 adrenergic receptor in living cells. Biochem. Biophys. Res. Commun. 2006, 343, 1191–1196. DOI: https://doi.org/10.1016/j.bbrc.2006.03.064
- Feng, J.; Zhang, C.; Lischinsky, J.E.; et al. A Genetically Encoded Fluorescent Sensor for Rapid and Specific In Vivo Detection of Norepinephrine. Neuron 2019, 102, 745–761. DOI: https://doi.org/10.1016/j.neuron.2019.02.037
- Muller, A.; Joseph, V.; Slesinger, P.A.; et al. Cell-based reporters reveal in vivo dynamics of dopamine and norepinephrine release in murine cortex. Nat. Methods 2014, 11, 1245–1252. DOI: https://doi.org/10.1038/nmeth.3151
- Surdo, N.C.; Berrera, M.; Koschinski, A.; et al. FRET biosensor uncovers cAMP nano-domains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 15031. DOI: https://doi.org/10.1038/ncomms15031
- Inagaki, H.K.; Ben-Tabou de-Leon, S.; Wong, A.M.; et al. Visualizing neuromodulation in vivo: TANGO-mapping of dopamine signaling reveals appetite control of sugar sensing. Cell 2012, 148, 583–595. DOI: https://doi.org/10.1016/j.cell.2011.12.022
- Caldwell, J.L.; Lee, I.J.; Ngo, L.; et al. Whole-heart multiparametric optical imaging reveals sex-dependent heterogeneity in cAMP signaling and repolarization kinetics. Sci. Adv. 2023, 9, eadd5799. DOI: https://doi.org/10.1126/sciadv.add5799
- Fazal, L.; Azibani, F.; Vodovar, N.; et al. Effects of biological sex on the pathophysiology of the heart. Br. J. Pharmacol. 2014, 71, 555–566. DOI: https://doi.org/10.1111/bph.12279
- Luczak, E.D.; Leinwand, L.A. Sex-based cardiac physiology. Annu. Rev. Physiol. 2009, 71, 1–18. DOI: https://doi.org/10.1146/annurev.physiol.010908.163156
- Yue, Q.; Wang, K.; Guan, M.; et al. Single-Vesicle Electrochemistry Reveals Sex Difference in Vesicular Storage and Release of Catecholamine. Angew. Chem. Int. Ed. Engl. 2022, 61, e202117596. DOI: https://doi.org/10.1002/anie.202117596
- Mitoff, P.R.; Gam, D.; Ivanov, J.; et al. Cardiac-specific sympathetic activation in men and women with and without heart failure. Heart 2011, 97, 382–387. DOI: https://doi.org/10.1136/hrt.2010.199760
- Cingolani, O.H.; Kaludercic, N.; Paolocci, N. Sexual dimorphism in cardiac norepinephrine spillover: A NET difference. Heart 2011, 97, 347–349. DOI: https://doi.org/10.1136/hrt.2010.217133
- Grimsby, J.; Toth, M.; Chen, K.; et al. Increased stress response and beta-phenylethylamine in MAOB-deficient mice. Nat. Genet. 1997, 17, 206–210. DOI: https://doi.org/10.1038/ng1097-206
- Jiang, H.; Xie, T.; Ramsden, D.B.; Ho, S.L. Human catechol-O-methyltransferase down-regulation by estradiol. Neuropharmacology 2003, 45, 1011–1018. DOI: https://doi.org/10.1016/S0028-3908(03)00286-7


