

Downloads
Download
Additional Files
Download - Supplemengary Material



This work is licensed under a Creative Commons Attribution 4.0 International License.
Article
Overexpressing Mitogen-activated Protein Kinase Kinase 7 (MKK7) Alleviates Endoplasmic Reticulum Stress-induced Cardiac Dysfunction during Pressure Overload induced Heart Failure
Tayyiba Azam 1, * , Min Zi 1, Susanne S. Hille 2,3, Hongyuan Zhang 1, Elizabeth J. Cartwright 1, Oliver J. Müller 2,3, and Xin Wang 1
1 Faculty of Biology, Medicine, and Health, University of Manchester, Oxford Road, M13 9PT, Manchester, UK
2 Department of Internal Medicine III, University of Kiel, 24105 Kiel, Germany
3 DZHK, German Centre for Cardiovascular Research, Partner Site Hamburg/Kiel/Lübeck, 20251 Hamburg, Germany
* Correspondence: Tayyiba.azam@manchester.ac.uk
Received: 12 June 2023
Accepted: 9 August 2023
Published: 27 December 2023
Abstract: Since its emergence as a cardiovascular epidemic in 1997, heart failure (HF) continues to be a significant challenge globally both economically and in clinical practice. As a consequence of the lack of effective treatments available, significant efforts have been devoted with the aim of identifying novel therapeutic strategies to treat HF. Loss of Mitogen-activated Protein Kinase Kinase 7 (MKK7) in the myocardium has previously been shown to underpin ventricular hypertrophy and HF progression. We demonstrated that adeno-associated virus 9 (AAV9) cardiac-specific overexpression of MKK7 therapeutically impeded cardiac dysfunction, interstitial fibrosis and cardiomyocytes apoptosis following 5-weeks pressure overload-induced HF. It was found that this was achieved, at least partly, by ameliorating Endoplasmic Reticulum (ER) stress. Mechanistically, in vivo and in vitro analysis revealed that although overexpression of MKK7 had no effect on PERK-ATF4 or ATF6 signalling pathways, the IRE1-XBP1 signalling pathway was preserved and a hindered increase in CHOP expression was observed. In conclusion, overexpression of MKK7 holds therapeutic potential in mitigating the pathological cardiac changes associated with HF progression.
Keywords:
Heart failure cardio-protection ER stress gene-therapy1. Introduction
Ventricular hypertrophy is undeniably a prerequisite for the development and progression of heart failure (HF). In the face of acute stress, the heart initially invokes an adaptive response to maintain cardiac function and normalize ventricular wall stress; however, chronic or severe insults can result in decompensatory maladaptive hypertrophy which is characterized by left ventricular dilation, interstitial fibrosis, cardiomyocyte apoptosis and a reduction in cardiac function [1], increasing the likelihood of heart failure development and progression [2]. Adult cardiomyocytes have limited regenerative ability and therefore rely on a vital balance in protein integrity [3]. Therefore, regulated endoplasmic reticulum (ER) response to changes in protein quality is paramount for optimum cellular function. Due to the dynamic nature of the ER, it is involved in a plethora of cellular processes including coordinating protein synthesis and folding [4], uptake and storage of intracellular calcium, and lipid and sterol biosynthesis [5].
Any alteration in the cell can overwhelm the ER protein folding capacity, resulting in an accumulation of misfolded protein within the lumen of the ER. Exposed hydrophobic regions of misfolded proteins can bind to other similar regions in other nearby proteins, leading to the formation of toxic aggregates, pre-amyloid oligomers and polypeptides. This can greatly affect cardiomyocyte function and survival [6]. As cardiomyocytes are post-mitotic cells and have limited regenerative capacity, it is imperative that protein homeostasis is maintained. ER stress has been associated with both physiological and pathological stress in the cardiovascular system such as myocardial ischaemia/reperfusion injury [7], diabetes [8], heart failure [9] pressure-overload stress [10] and dilated cardiomyopathy [11]. The cell adjusts to any fluctuations and discrepancies in protein folding via a dynamic integration of adaptive mechanisms, known collectively as the unfolded protein response (UPR). By buffering ER stress, the UPR orchestrates the recovery of ER function. The intensity and duration of the ER stress stimuli govern the UPR. Acute ER stress can be overcome ensuring the survival of the cells. However, during prolonged or severe ER stress the damaged cell is eliminated through apoptosis [12]. The UPR is a three-armed stress responsive signalling pathway consisting of activating inositol-requiring enzyme 1 (IRE1), protein kinase-like ER kinase (PERK) and activating transcription factor 6 (ATF6). These pathways are activated by specific mechanisms and govern different downstream signalling proteins.
Mitogen-activated protein kinase kinase 7 (MKK7) is a stress-activated kinase that is a member of the mitogen-activated protein kinase kinase family. It is primarily involved in the activation of c-Jun N-terminal kinase (JNK). In the context of the heart, it has previously been described to play a cardio-protective role in the heart. Cardiac loss of MKK7 renders the heart vulnerable to cardiac dysfunction and pathological ventricular remodelling [13]. Moreover, it has also been found that cardiomyocyte-specific deprivation of MKK7 promotes arrhythmia development and that transgenic mice overexpressing MKK7 are resistant to these pathological changes [14]. However, it is unknown whether therapeutically maintaining MKK7 expression can ameliorate cardiac dysfunction.
In this study, we seek to ascertain that cardiac MKK7 overexpression holds therapeutic value in limiting HF progression. We demonstrated that cardiac MKK7 expression is downregulated in the failing mouse heart; however, therapeutically maintaining MKK7 expression mitigated cardiac dysfunction, pathological remodelling, fibrosis, and cell death. Mechanistically, it was identified using both in vivo and in vitro models, that upregulation of cardiomyocyte-specific MKK7 alleviated ER stress likely through activation of the IRE1-XBP1 signalling pathway. In addition, hindered CHOP expression was also observed following MKK7 overexpression, suggesting limited ER stress-mediated cell death. In summary, we have provided proof-of-concept evidence of a novel cardio-protective mechanism whereby MKK7 relieves ER stress in pressure-overload-induced HF.
2. Methods
All laboratory mice used in this study were maintained in a pathogen-free facility at the University of Manchester. Animal studies were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and were approved by the University of Manchester Ethics Committee.
2.1. Transverse Aortic Construction
8‒10 week-old male C57BL/6J mice underwent transverse aortic constriction as previously described [8]. Briefly, after the mice were anaesthetised using isoflurane (Isothesia, Henry Schein) and ventilated, the transverse aorta between the right innominate and left common carotid arteries was subjected to a 27-guage constriction by a 7-0 Prolene suture. Buprenorphine (0.1 mg/kg) was administrated for analgesia. A similar procedure was performed on sham mice; however, the suture was not tied.
2.2. Echocardiography
For cardiac function evaluation, mice were anaesthetised with 1.5% isoflurane mixed with 100% oxygen at 1.5 L/min rate. The Acuson Sequoia C256 system (Siemens) and Vevo 770 system (Visualsonics) were used to obtain transthoracic M-Mode and pulse wave Doppler ultrasound images. Parameters such as fractional shortening (FS%), left ventricle chamber and wall dimensions, diastolic function (IVRT, E/A) parameters, ejection fraction (EF%), left ventricular (LV) mass and relative wall thickness were measured or calculated as described previously [10]. To confirm the presence of transverse aortic constriction (TAC), the distal transverse aortic flow velocity was calculated using pulse wave doppler and the modified Bernoulli equation (Pressure gradient = 4 × Velocity2).
2.3. Construction of Adeno-associated Virus-MKK7 (AAV9-TnT-MKK7)
The pSSV9-TnT-eGFP was modified by replacing GFP with Flag-tagged human MKK7 cDNA (DU48691, MRCPPU reagents). To obtain the recombinant AAV9-MKK7 virus and the control AAV9-eGFP virus, HEK293T cells were co-transfected with an adenoviral helper plasmid (pDG∆VP), pAAV2-9 Rep-Cap plasmid (p5E18-VD2/9) and the adeno-associated virus genome plasmids pSSV9-TnT-MKK7 and pSSV9-TnT-eGFP, respectively. Afterwards, recombinant AAV9-MKK7 was purified by discontinuous iodixanol gradient and titrated by quantitative polymerase chain reaction (qPCR) relative to a standard curve. To overexpress MKK7, 1 × 1011 genomic particles were administrated by intravenous injection. AAV9-GFP was injected as the control.
2.4. Histology
Isolated hearts were immediately fixed in 4% paraformaldehyde for 5 h at 4 °C. Next, they were washed in PBS for 30 min before being dehydrated in 50%, 70%, 90% and 100% ethanol for 2 h each. The hearts were subsequently, cleared in xylene overnight, and embedded in paraffin. Blocks were sectioned into 5 μm thickness using Microtome (Leica). Sections were dewaxed and rehydrated prior to histological staining.
2.5. Haematoxylin & Eosin (H&E) Staining
Haematoxylin and eosin staining was used to evaluate cardiomyocyte cross-sectional area. Sections were stained with Harris Haematoxylin (LAMB/230, RA Lamb Dry Chemical Stains) for 5 min, followed by differentiated in 1% HCL in 70% ethanol solution for 10 s. After washing in warm running water for 5 min, Alcoholic Eosin (6766007, ThermoScientific) was added to the sections for 1 min. After rinsing twice in ddH2O, sections were dehydrated in 50%, 70%, and 100% ethanol for 5 min each, then cleared in xylene for 20 min. Samples were mounted with Eukitt (03989, Sigma). Slides were imaged using 3D Histech Panoramic 250 Slide-scanner. For quantification, ventricular cross-sectional area of 100 randomly selected cardiomyoyctes, for each heart, was analysed using ImageJ.
2.6. Masson’s Trichrome Staining
Masson’s trichrome staining was used to examine interstitial fibrosis. Firstly, sections were fixed with Bouin’s fixative (HT10132, Sigma) at room temperature for 2 h. Next, sections were incubated with Harris’s Haematoxylin (LAMB/230, RA Lamb Dry Chemical Stains) for 5 min, before undergoing differentiated in 1% HCL in 70% ethanol solution for 10 s. After washing in warm running water for 5 min, red solutuin (HT151, Sigma) was added for 5 min. After washing in cold ddH2O, sections were treated with 2.5% phosphomolybdic acid (HT153, Sigma) for 15 min. Subsequently, sections were stained with aniline blue solution (B8563, Sigma) for 5 min followed by 1% acetic acid for 1 min. Finally, sections were dehydrated in 50%, 70%, and 100% ethanol for 5 min each, then cleared in xylene for 20 min. Slides were mounted with Eukitt (03989, Sigma). Slides were imaged using 3D Histech Panoramic 250 Slide-scanner. The color threshold function on ImageJ was used to calculate the areas of fibrosis from 10 fields per sample.
2.7. TUNEL Assay
TUNEL assay was performed to detect apoptotic cardiomyocytes using the DAPI, TUNEL (In situ Cell Death Detection kit, Roche), and actinin (A7811, Sigma), following the manufacturer’s instructions. The primary α-actinin antibody was incubated overnight at 4 °C. The next day, secondary anti-mouse IgG Alexa-594 conjugate antibody (715-585-150, Jackson ImmunoResearch) for 2 h at room temperature. Sections were mounted with DAPI-containing Vectashield (H-1200, Vector Laboratories). Slides were imaged using DAPI, FITC and Texas Red filter channels. A total of 10,000 cardiomyocytes from random fields of the heart were analysed. Images were obtained by Zeiss Axioplan2 microscope and analysed by ImageJ software.
2.8. Electron Microscopy
For analysis of changes in endoplasmic reticulum structure, transmission electron microscopy was performed. Freshly isolated heart samples were fixed overnight in 0.1 mol/L HEPES buffer (pH 7.2) containing 2.5% glutaraldehyde and 4% paraformaldehyde. Next, tissues were processed in 0.1 mol/L Cacodylate Buffer (pH 7.2) with 1.5% potassium ferrocyanide and 0.1% osmium tetroxide for 1 h, before being incubated with 0.1 mol/L Cacodylate Buffer (pH 7.2) with 1% tannic acid for 1 h and finally 1% uranyl acetate or a further hour. Next, samples were dehydrated in ethanol and embedded in TAAB 812 resin before being polymerised at 60 °C for 24 h. Finally, the sections were cut with a Reichert Ultracut Ultramicrotome and examined using the Talos L120C transmission electron microscope at 100 kV accelerating voltage. Images were taken with Gatan Orius SC1000 CCD camera.
2.9. Immunoblot
Total protein was obtained with Triton lysis buffer (137 mmol/L NaCl, 20 mmol/L Tris, 0.1% w/v SDS, 2 mmol/L EDTA, 10% v/v glycerol, 1% Triton-X, 25 mmol/L glycerophosphate, 1 mmol/L Na3VO4, 1 mmol/L, 10 mmol/L NAM, 1 μmol/L TSA, 1x protein cocktail inhibitor, pH7.4). Lysates were cleared by centrifugation at 14,000 g for 20 min. Protein concentration was determined by Bio-Rad protein assay. Immunoblot analysis was performed on 30 µg protein extracts with antibodies against MKK7 (4172, Cell Signalling), Flag (MA1-91878, Thermofisher), phospho-MKK7 (4171, Cell Signalling), JNK (9252, Cell Signalling), p38 (9212, Cell Signalling), ERK1/2 (9102, Cell Signalling), MKK4 (9152, Cell Signalling), GRP78 (ab21685, Abcam), IRE1 (ab37073, Abcam), phospho-IRE (ab48187, Abcam), XBP1s (24868-1-AP, Proteintech), ATF6 (ab37149, Abcam), PERK (3192, Cell Signalling), phospho-PERK (3179, Cell Signalling), eIF2α (9722, Cell Signalling), phospho-eIF2α (9721, Cell Signalling), ATF4 (10835-1-AP, Proteintech), CHOP (2895, Cell Signalling) and G-Beta (166123, Santa Cruz Biotechnology). Secondary antibodies anti-rabbit (7074, Cell Signalling) or anti-mouse (7076, Cell Signalling) HRP conjugates were used. Immune-complexes were detected using Amersham ECL Prime/Select detection reagents (RPN2232/RPN2235, Amersham) and the ChemiDoc MP System (BioRad).
2.10. Quantitative Real-time Polymerase Chain Reaction (qPCR)
Total RNA from cells and ventricular tissues was extracted using Trizol (Invitrogen). Samples were treated with DNAse (DNA-free Removal Kit, Invitrogen) to eliminate DNA contamination. RNA was converted into cDNA using Lunascript (NEB3010, New England Biolabs). All primers were purchased from Qiagen and qPCRs were conducted using SYBR Select PCR Master Mix (4472908, Applied Biosystems) following the manufacturer’s instructions. qPCR reactions were run in the Step One Plus PCR System (Applied Biosystems), and fold change was calculated by the comparative Ct (ΔΔCt) method. mRNA levels were normalized to 18S expression.
2.11. H9C2 Cell Culture
H9C2 cells were obtained from the European Collection of Authenticated Cell cultures (88092904, Sigma-Aldrich). Cells were maintained in DMEM (11966-025, Gibco) containing 1 g/L glucose and 10% FBS. To mimic the effects of TAC-induced pressure-overload stress in vitro, cells were exposed to isoprenaline (I5627, Sigma). Time-points are stated in the corresponding figures.
2.12. Adenoviral Production and Infection
The human MKK7 cDNA sequence was subcloned into pENTR11 plasmid (A10467, Thermofisher Scientifics). The sequence was then recombined to pAd/CMV/V5-DESTTM (Invitrogen). DNA plasmids were digested with PacI (New England Biolabs). This restriction endonuclease exposed two inverted terminal repeats (ITRs) to aid virus uptake, replication and packaging in cells. Human embryonic kidney 293T (HEK293T) cells were transfected with the purified plasmids. Cells were harvested once 80% cytopathological effect (CPE) was observed. Three cycles of freeze and thaw were performed to release the virus particles. Centrifugation in gradient caesium chloride (CsCl) (1.33 g/mL and 1.45 g/mL) was carried out and the band containing the adenovirus was removed and purified using a dialysis tubing (pore size 24 Angstrom, Medicell). Adenovirus infection was induced at 25 multiplicity of infection (MOI).
2.13. Proteostat Staining
Coverslips were sterilized for 30 min in 70% IMS at room temperature, before being washed with PBS and placed into a 24 well plate. H9C2 cells were counted using the Trypan Blue assay according to the standard protocol and seeded onto coverslips at a density of 4 × 104 cells per well. Cells were incubated and allowed to grow until at least 70% confluency was achieved. Next, cells were infected with either Ad-GFP or Ad-MKK7 before being subjected to 48 hours isoprenaline (100 µmol/L). Then, coverslips were washed with PBS and fixed with 4% paraformaldehyde for 20 min. Subsequently, Proteostat staining was performed according to the manufacturer’s instructions to examine protein aggregation (ENZ-51023).
2.14. Data Analysis
Data are presented as mean ± SEM and analysed using one-way or two-way ANOVA followed by post hoc tests where appropriate. Comparisons between two groups were performed using Student’s t test. For data with skewed distribution, non-parametric tests were used. Statistical analysis was performed using the GraphPad Prism 9 software and p < 0.05 was considered statistically significant.
3. Results
3.1. MKK7 is Down-regulated in Cardiac Pathological Stress
In order to investigate the role of MKK7 in the myocardium, we first assessed changes in MKK7 expression following transverse aortic constriction. To do so, we subjected male C57BL/6J mice to either one-week or 5-weeks pressure-overload stimulation, which represents acute or prolonged stress, respectively. We found that MKK7 phosphorylation was increased under acute stimulation; however, MKK7 expression was maintained. In contrast, both MKK7 expression and phosphorylation were decreased by pressure overload stress after 5 weeks (Figure 1A). Next, an in vitro model of isoprenaline, commonly used to mimic the conditions of hypertrophic growth was utilised. In line with observations from the mouse myocardium, we observed an oscillating pattern of MKK7 phosphorylation. Total MKK7 expression was maintained during short-term stress; however, it was decreased following prolonged and sustained isoprenaline-induced stress (Figure 1B). Lower doses of isoprenaline did not elicit the same response (Supplementary Figure S1). Together, these results demonstrated decreased MKK7 expression in the failing heart.
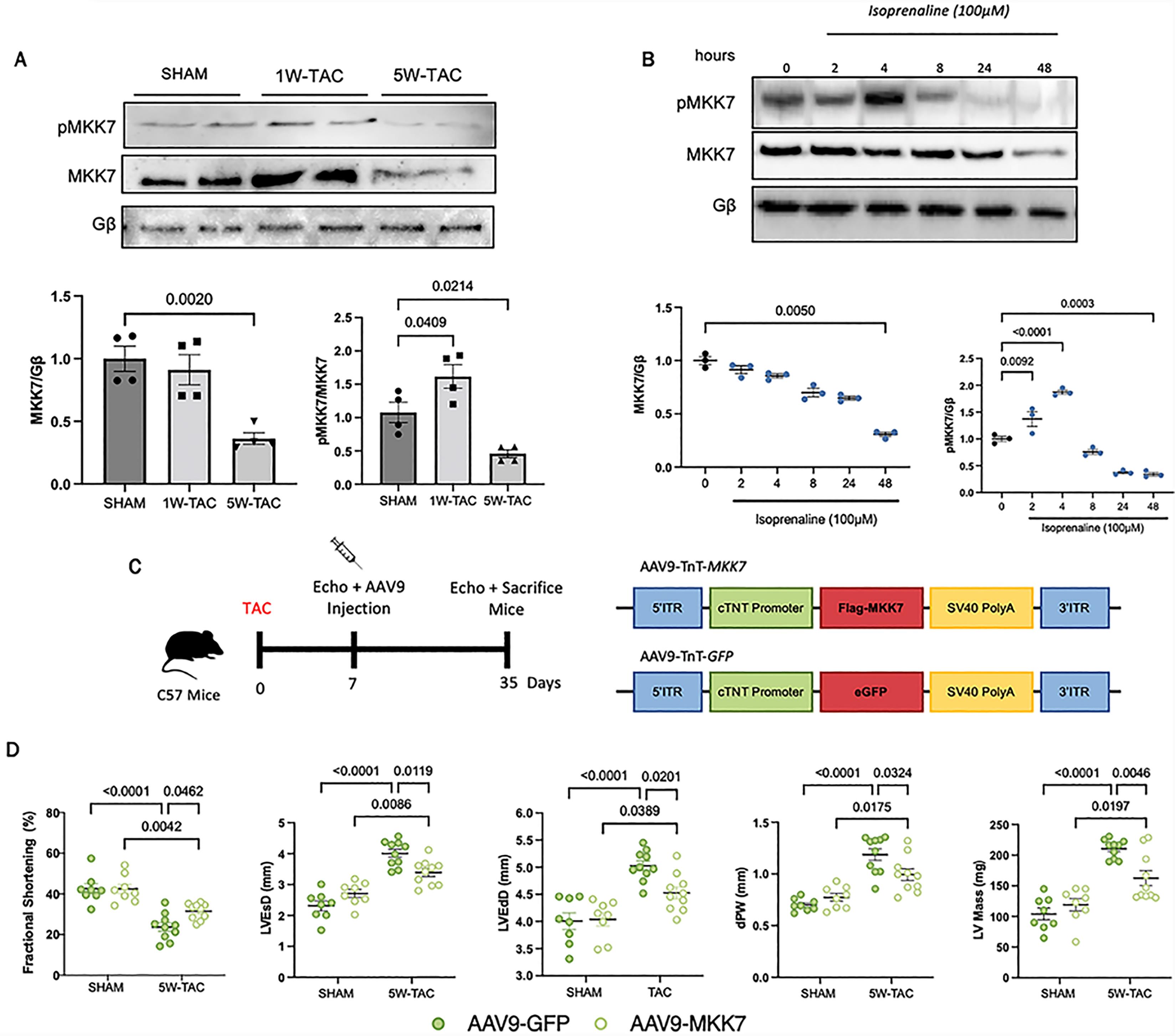
Figure 1. Downregulated MKK7 expression following pressure-overload stress and isoprenaline treatment. A: Immunoblotting analysis of total and phosphorylated MKK7 in C57BL/6J hearts after SHAM, 1W-TAC or 5W-TAC (N = 4); B: Immunoblotting analyses of H9C2 cells treated with 100 µmol/L isoprenaline for different timepoints (N = 3); C: Construction of AAV9-TnT-eGFP and AAV9-TnT-MKK7 and experimental overview of pressure-overload induced stress on AAV9-TnT-eGFP and AAV9-TnT-MKK7 injected male C57BL/6J mice; D: Fractional shortening (FS%), left ventricular end-systolic diameter, left ventricular end-diastolic diameter, Diastolic posterior wall thickness and left ventricular mass (N = 8–10). Data presented as means ± SEM.
3.2. MKK7 Prevents Pathological Ventricular Remodelling and Heart Failure
Downregulated MKK7 in the failing heart suggests that MKK7 expression coincides with the transition from compensatory to decompensatory ventricular remodelling and the rationale that MKK7 may play a cardioprotective role in the heart by ameliorating cardiac dysfunction. To examine whether maintaining MKK7 expression could provide therapeutic benefit, we first subjected 8–10 week-old C57BL/6J male mice to transverse aortic constriction (TAC). One week later we induced cardiomyocyte-specific overexpression of MKK7 by injecting the mice with AAV9-mediated delivery of troponin T (TnT) promoter-driven human MKK7 (AAV9-TnT-MKK7, 1 × 1011 virus particles). AAV9-TnT-eGFP was used as the control (Figure 1C–1D). Characterisation of AAV9-TnT-MKK7 injected mice demonstrated cardiomyocyte-specific MKK7 overexpression (Supplementary Figure S2). After 5 weeks pressure-overload stimulation cardiac function and morphological assessment was performed. Echocardiographic analysis found that in contrast to AAV9-TnT-eGFP TAC mice, which developed cardiac dysfunction, AAV9-TnT-MKK7 mice manifested resilience to impaired cardiac contractility and dysfunction compared to control mice, exemplified by a higher fractional shortening (FS %) and a blunted increase in ventricular wall mass. They also exhibited significantly reduced left ventricular end-systolic diameter (LVEsD), left ventricular end-diastolic diameter (LVEdD), posterior wall thickness (dPW) and left ventricular mass (Figure 1E). With regard to pathological hypertrophic remodelling, enlargement of the cardiomyocyte cross-sectional area was observed in AAV9-TnT-eGFP injected mice subjected to 5-week pressure-overload stress. In contrast, AAV9-TnT-MKK7 injected mice demonstrated a blunted increase (Figure 2A–2B). A similar response was observed upon analysis of the mRNA level of key hypertrophic markers. The transcript expression levels of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP) and myosin heavy chain 7 (Myh7) were shown to be significantly lower in AAV9-TnT-MKK7 TAC mice compared to AAV9-TnT-eGFP TAC-subjected mice (Figure 2C). Following 5-weeks TAC treatment, there was a greater increase in the ratio of heart weight/tibia length in AAV9-TnT-eGFP mice compared to AAV9-TnT-MKK7 mice. In addition, AAV9-TnT-eGFP mice also experienced increased lung weight/tibia length indicating pulmonary congestion as a result of cardiac dysfunction (Figure 2D). Furthermore, reduced interstitial fibrosis was evident in AAV9-TnT-MKK7 mice following 5-week TAC compared to AAV9-TnT-eGFP TAC-subjected mice (Figure 2E). Increased cardiomyocyte apoptosis is a key response following pressure-overload stress. Therefore, TUNEL staining was performed to identify differences in cardiomyocyte apoptosis between AAV9-TnT-eGFP and AAV9-TnT-MKK7 mice following TAC. As seen in 2G-2H, AAV9-TnT-eGFP mice cardiac tissue had elevated levels of TUNEL positive cells following 5-week TAC, whereas this response was attenuated in AAV9-TnT-MKK7 mice. Collectively, these data demonstrated the functional benefit in maintaining MKK7 expression in protecting the heart from heart failure development following chronic pressure-overload stimulation.

Figure 2. MKK7 overexpression mitigated pathological ventricular remodelling, interstitial fibrosis and cardiomyocyte apoptosis. A and B: H&E staining analysis of cardiomyocyte cross-sectional area. Scale bar: 50 µmol/L. (N = 8–10); C: Relative mRNA expression of ANP, BNP and Myh7 (N = 5–6); D: Heart weight/tibia length and lung weight/tibia length (N = 8–10); E and F: Masson’s Trichrome staining and analysis of interstitial fibrosis. Scale bar: 50 µmol/L. (N = 8–10); G: TUNEL staining of the hearts. Scale bar: 50 µmol/L. (N = 8–10). Data presented as means ± SEM.
3.3. MKK7 Mitigates ER Stress by Activating the IRE1-XBP1 Pathway
Next, we attempted to gain insight into how MKK7 prevents cardiac dysfunction. Multiple lines of evidence have shown that an accumulation of misfolded proteins underlies the pathophysiological response during various cardiac diseases, including pressure-overload stress. Consequently, we first examined changes in the unfolded protein response markers. Although total IRE1 levels were comparable between all groups of mice, AAV9-TnT-eGFP mice showed a blunted increase in phosphorylated IRE1 levels in comparison to AAV9-TnT-eGFP mice following 5-week TAC. Furthermore, XBP1 splicing levels were significantly higher in AAV9-TnT-MKK7 mice after TAC indicating increased IRE1/XBP1 signalling. The expression level of phosphorylated PERK, phosphorylated eIF2α, and ATF4 increased after pressure-overload stress, however the levels were comparable between AAV9-TnT-eGFP and AAV9-TnT-MKK7 mice. There was no significant difference in the total PERK, eIF2α and ATF6 between SHAM and 5-week TAC mice. Also, protein expression of CHOP was enhanced in control mice but less so in AAV9-TnT-MKK7 mice (Figure 3A–3B). Furthermore, analysis of the transcript level of components of the ER-associated degradation pathway including, Hrd1, Edem1, Derlin3 and UFD1 were upregulated in AAV9-TnT-MKK7 mice in response to pressure-overload stress (Figure 3C). In addition, ultrastructural analysis via transmission electron microscopy demonstrated slight expansion of the ER lumen, a response that was less prominent in AAV9-TnT-MKK7 TAC mice (Figure 3D). Therefore, the data obtained revealed that MKK7 overexpression maintained IRE1-XBP1 signalling to mitigate pathological stress-induced cardiac dysfunction.

Figure 3. Overexpression of MKK7 ameliorated ER stress by upregulation of IRE1-XBP1 signalling. A and B: Representative immunoblot images and quantification of UPR markers in SHAM or 5wk-TAC hearts infected with either AAV9-TnT-eGFP or AAV9-TnT-MKK7 (N = 5–8). C: Relative mRNA expression of components of the ERAD and UPR pathway in heart lysates from TAC AAV9-TnT-eGFP and TAC or AAV9-TnT-MKK7 mice (N = 5). Data presented as means ± SEM; D: Transmission electron microscopic images of hearts subjected to 5-weeks TAC from AAV9-TnT-eGFP or AAV9-TnT-MKK7 mice. Scale bar = 500 nm.
3.4. MKK7 Suppresses ER Stress during Isoprenaline Treatment
An in vitro model of MKK7 overexpression, via adenovirus infection, was utilised to confirm MKK7 regulation of the UPR. Firstly, cells were treated with isoprenaline for various timepoints to examine changes in the UPR. Although total IRE1 were maintained throughout the timepoints, the expression of phosphorylated IRE1 and spliced-XBP1 demonstrated an oscillating pattern. The level of phosphorylated IRE1 and spliced XBP1 reached peak expression at 4th h after isoprenaline treatment before returning back to baseline. The expression of ATF4 displayed a similar pattern, whereby it reached its peak at 4 h before returning to levels similar to the vehicle control. ATF6 levels remained constant through the isoprenaline treatment. The pro-apoptotic factor, CHOP, increased expression at 8 h and remained increasing until 48 h (Figure 4A). In order to investigate the effect of MKK7 on ER stress in vitro, cells were infected with either Ad-MKK7 or Ad-GFP (Supplementary Figure S3). After 48 h, changes in the expression of key ER stress components as this was considered at the timepoint were MKK7 expression decreased (Figure 1B). Overexpression of MKK7 partly rescued IRE1-XBP1 signalling and dampened CHOP expression (Figure 4B). There was also prominent protein aggregation in Ad-GFP infected cells treated with isoprenaline compared to Ad-MKK7 infected cells (Figure 4C).
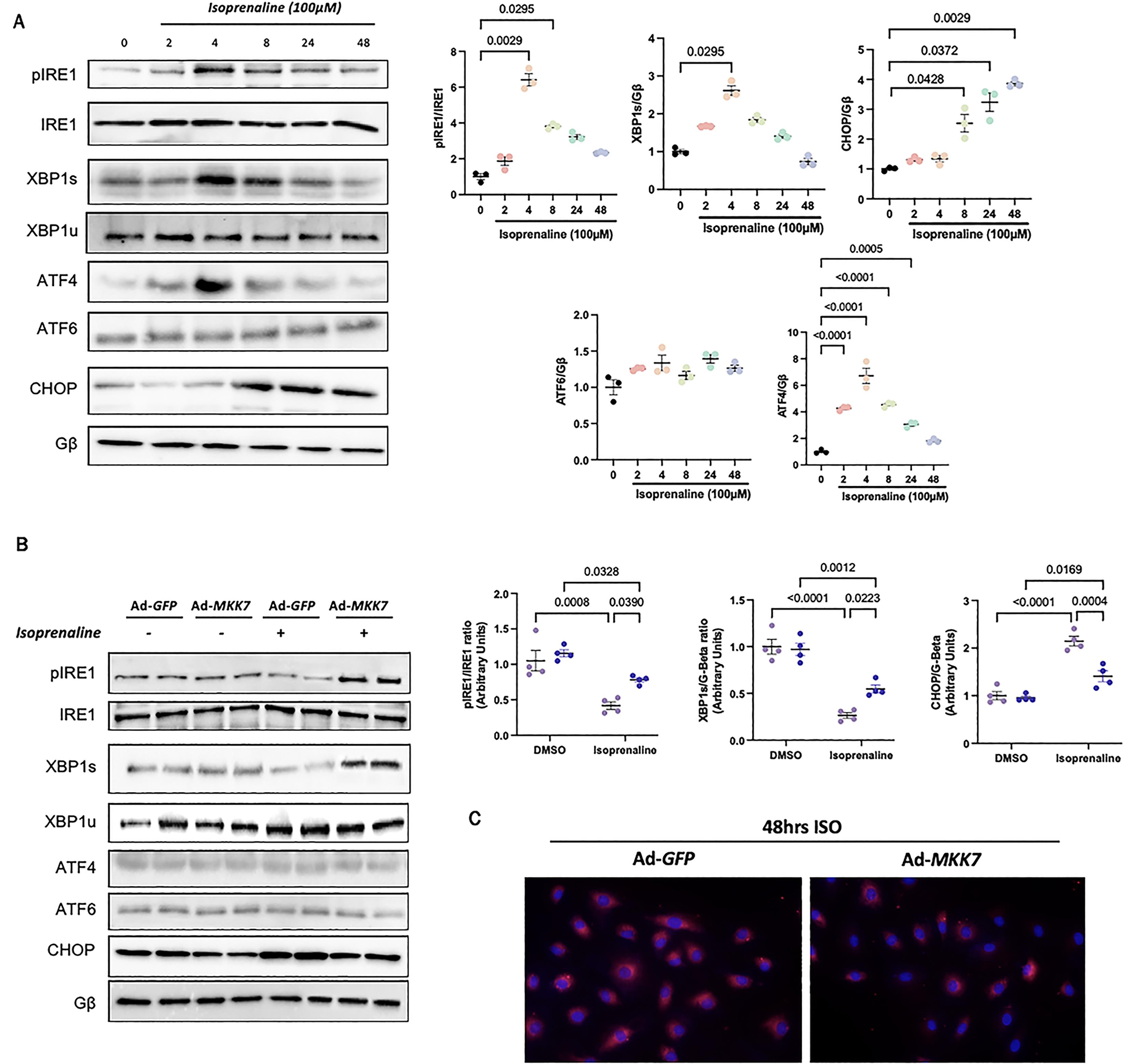
Figure 4. In vitro evidence for MKK7 alleviation of ER stress during isoprenaline treatment. A: Representative immunoblot images and quantification of UPR markers in H9C2 cells subjected to 100 µmol/L isoprenaline treatment for different time points (N = 3); B: Representative immunoblot images and quantification of IRE1, XBP1 and CHOP in H9C2 cells infected with Ad-GFP or Ad-MKK7 subjected to 100 µmol/L isoprenaline treatment or DMSO for 48 h (N = 4); C: Proteostat staining of H9C2 cells infected with Ad-GFP or Ad-MKK7 subjected to 100 µmol/L isoprenaline treatment for 48 h (N = 4).
4. Discussion
Led by the observation that MKK7 expression is reduced in the failing heart, this study found that maintaining MKK7 expression (1) prevented cardiac dysfunction and pathological ventricular remodelling, (2) mitigated myocardial fibrosis and cell death and (3) alleviated ER stress by preserving IRE1-XBP1 signalling.
4.1. MKK7 Overexpression Suppressed Pathological Cardiac Hypertrophy
Pressure overload stress in the heart increases left ventricular afterload, followed by a compensatory increase in left ventricular hypertrophy. This is considered an adaptive response and initially cardiac function is maintained. However, sustained pressure-overload promotes the progression from compensatory to decompensatory maladaptive hypertrophy. This is characterised by left ventricular dilation and a reduction in cardiac function [15]. This study found that the expression of MKK7 coincided with the transition from compensatory to decompensatory ventricular remodelling. The expression of total MKK7 decreased following long-term (5-week) pressure-overload stress. MKK7 phosphorylation increased following short-term pressure-overload stress (1-week) but long-term stress suppressed its expression. This indicates that it may play a vital role in HF progression. MKK7 expression decreased following long-term isoprenaline treatment further reiterating that MKK7 expression is sustained in short-term stress but is degraded upon long-term stress. Lower doses of isoprenaline did not inflict any changes in MKK7 abundance or activation further demonstrating that MKK7 is a stress-activated kinase that is activated upon long-term excessive stress. However, the mechanism whereby MKK7 is downregulated in the stressed heart and cardiomyocytes after sustained stress remained elusive.
This study aimed to investigate the therapeutic potential of MKK7 during pressure overload stress. Consequently, MKK7 overexpression was established one-week after the initiation of TAC stress. As the first week is known to involve compensatory changes in the heart to maintain cardiac function, MKK7 was given after this stage with the aim to prolong the compensatory changes and mitigate the decompensatory effects in the heart. Research investigating MKK7’s function in the heart has contradicting findings, indicating it may play a dichotomous role and that further research is required to establish the true role of MKK7 in the heart. We and others have revealed the cardio-protective role of MKK7 in the heart in response to pathological stress [13‒16]. However, a number of other studies have suggested that MKK7 may play a detrimental role in the heart [17,18]. These discrepancies are possibly attributed to the use of different MKK7 isoforms and the use of constituently active MKK7.
4.2. MKK7 Overexpression Mitigated ER Stress
Despite discrepancies in disease phenotypes, impairment of the UPR signalling underlies the pathophysiology of numerous cardiovascular diseases, including diabetes [19], cardiac hypertrophy [20] and heart failure [21]. The UPR not only regulates protein folding but also lipid metabolism, redox homeostasis, autophagy and glycosylation, implying that it is involved in a wide range of processes [22]. During cardiac hypertrophy, there is increased demand for protein synthesis requiring activation of the UPR to maintain protein homeostasis particularly during ER stress. Chronic ER stress negatively impacts cardiac function, damaging the myocardium and leading to HF [23]. Targeting protein quality control to sustain protein homeostasis holds the potential to alleviate cardiac dysfunction and promote health [24].
MKK7 overexpression was able to maintain ER homeostasis as assessed by UPR markers in gain-of-function in vivo and in vitro models. Our data demonstrated that MKK7-mediated signalling led to IRE1 phosphorylation resulting in the activation of the transcription factor XBP1s. This in turn upregulated components of the ERAD pathway and chaperone proteins to alleviate ER stress. Previous studies have identified the vital role of IRE1-XBP1 signalling in the failing heart. XBP1 splicing expression is decreased in both human and rodent cardiac myocardium tissue under heart failure [25]. Using AAV9 carrying short hairpin RNA against XBP1, Duan et al. [26] demonstrated that loss of XBP1 instigated aberrant angiogenesis, and promoted the progression of maladaptive cardiac hypertrophy, exacerbated isoprenaline-induced cardiac dysfunction. Duan et al. [26] and Wang et al. [25] showed using both loss-of-function and gain-of-function models the involvement of XBP1 expression in heart failure. Cardiac-restricted XBP1 deficiency exacerbates HF progression following pressure overload stress whereas cardiomyocyte-specific XBP1 overexpression prevented cardiac dysfunction [25]. In addition, overexpression of cardiomyocyte-restricted IRE1α conferred cardioprotection evident by preserved cardiac function, decreased myocardial fibrosis, blunted inflammatory response and increased UPR signalling [27]. Akin to the observations upon TAC-stress, MKK7 overexpression preserved IRE1-XBP1 expression in H9C2 cells subjected to isoprenaline stress and reduced protein aggregation. In addition, increased CHOP expression during pressure-overload stress was hindered by MKK7 overexpression, suggesting reduced cardiomyocyte cell death. CHOP is a transcription factor that regulates ER-mediated apoptotic cell death in various cells, not only cardiomyocytes [28]. It has previously been demonstrated that the level of CHOP expression is increased in the human failing heart [29]. Similarly, this study found that the level of CHOP expression increased in the myocardium of mice subjected to five-week TAC, coinciding with increased cardiomyocyte apoptosis which was examined using TUNEL staining.
4.3. Limited Myocardial Fibrosis with MKK7 Overexpression
Another observation unveiled by this study was that MKK7 overexpression suppressed excessive myocardial fibrosis in TAC conditions. Interstitial fibrosis is a hallmark feature during pathological ventricular remodelling. A well-known fibrogenic growth factor involved in cardiac fibrosis is TGF-β [30]. It is a pleiotropic mediator that has broad ranging functions including differentiation, proliferation and migration. TGF-β can induce the transformation, promote extracellular matrix component deposition [31], and prevent the degradation of the extracellular matrix by modulating the levels of tissue inhibitor of metalloproteinases (TIMPs) and plasminogen activator inhibitor 1 (PAI-1) [32]. This cytokine is involved in inhibiting collagen degradation and increasing collagen synthesis. Previously in a model of pressure overload stress, Liu et al. [13] suggested that MKK7 is a negative regulator of TGF-β. It is therefore plausible to propose that overexpression of MKK7 is likely responsible for suppressed TGF-β expression. However, further research is warranted to confirm this finding.
4.4. Upstream MKK7 Activators
Several upstream kinases have been discovered to activate MKK7 in the heart. Disrupted mitogen-activated protein kinase kinase kinase 1 (MAP3K1) expression in cardiomyocytes, predisposes mice to ventricular dilation and reduced cardiac function following pressure-overload stress, suggesting a cardioprotective. MAP3K1 was found to mitigate cardiac apoptosis and inflammation, and modulate JNK expression to safeguard the heart against heart failure progression following pressure-overload stress [33]. Similarly, it was found that mixed-linage kinase 3 (MLK3) regulates the activation of JNK in response to pressure-overload stress, revealing a novel MLK3-JNK signalling axis that protects the heart. Cardiomyocyte-restricted deletion of MLK3 resulted in baseline cardiac hypertrophy but cardiac function was preserved. However, following pressure-overload stress, mice developed significant cardiac dysfunction and hypertrophy [34]. The finding in these studies are comparable with the results from our study, whereby the MKK7 signalling axis plays a cardioprotective role in the setting of pressure-overload stress.
4.5. Limitations
This study found that long-term pathological stress in the form of pressure-overload stress resulted in the downregulation of the IRE1-XBP1 signalling cascade. MKK7 overexpression can preserve this signalling cascade; however, further examination of MKK7 confering preservation of the IRE1-XBP1 signalling pathway remains elusive.
5. Conclusion
We uncovered the cardioprotective role of maintaining MKK7 during TAC-induced pressure overload stress. This study presents, for the first time, evidence linking MKK7-mediated regulation of ER stress following pathological stress. MKK7 was able to regulate ER homeostasis to sustain protein quality control and mitigate pathological cardiac remodelling such as cardiac remodelling, fibrosis and cardiomyocyte cell death. Together this leads to a significant improvement in cardiac function following the onset of stress. Future work identifying modes of maintaining MKK7 expression in the heart is required for the development of novel strategies to prevent and treat heart failure.
Supplementary Materials: The following supporting information can be downloaded at: https://www. sciltp. com/ journals/ijddp/2023/4/313/196, Figure S1: Examina-tion of MKK7 abundance and activation following isoprenaline treatment in H9C2. Immunoblots and analyses of cells treated with various concentration of isoprenaline for different timepoints A 10 μM B 50 μM. N = 3. All data presented as mean ± SEM following Kruskal-Wallis Test. Figure S2: Immunoblot analysis of cardiac-specific overexpression of MKK7 in mice injected with AAV9-TnT-MKK7. A: Schematic diagram of constructed AAV9-TnT-MKK7 virus; B: Immunoblot confirming MKK7 overexpression upon constructed AAV9-TnT-MKK7 injection in the hearts of C57BL/6J mice; C: Immunoblotting confirming no overexpression or change in MKK7 expression in the liver, skeletal muscle or brain of AAV9-TnT-MKK7 injected mice. Figure S3: MKK7 overexpression in H9C2 cells. A: Schematic diagram of Ad-MKK7 adenoviral vector; B: Immunoblot and quantification confirming overexpression of MKK7 in H9C2 cells infected with Ad-MKK7 48 hours after infection.
Author Contributions: T.A designed and carried out experiments, analysed and interpreted data, and wrote the manuscript. M.Z. performed TAC surgery. H.Z. acquired various data. S.H. & O.M. produced AAV9-TnT-MKK7. E.J.C. designed animal works and reviews the study. X.W. conceptualized the project, designed experiments and interpreted results.
Funding: This study was supported by the British Heart Foundation (FS/19/39/34447) and a professorship from the German Centre for Cardiovascular Research (81Z0700201 to O.J. Müller).
Institutional Review Board Statement: The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of the University of Manchester (project license No. P2A97F3D 2021).
Informed Consent Statement: Not applicable.
Data Availability Statement: Not applicable.
Acknowledgments: The authors wish to thank Roger Meadows, Steven Marsden, Peter March, and Darren Thomson (Bioimaging facility, University of Manchester) for technical training on microscopes; in addition to Aleksandr Mironov FBMH EM Core Facility (RRID:SCR_021147) for assistance and the Wellcome Trust for equipment grant support to the EM Facility (University of Manchester). The Histology Facility equipment used in this study was purchased with grants from The University of Manchester Strategic Fund.
Conflicts of Interest: All authors have declared that no conflict of interest exists.
References
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. DOI: https://doi.org/10.1038/s41569-018-0007-y
- Riehle, C.; Bauersachs, J. Small animal models of heart failure. Cardiovasc. Res. 2019, 115, 1838–1849. DOI: https://doi.org/10.1093/cvr/cvz161
- Liu, M.; Liu, H.; Parthiban, P.; et al. Inhibition of the unfolded protein response reduces arrhythmia risk after myocardial infarction. J. Clin. Invest. 2021, 131, e147836 . DOI: https://doi.org/10.1172/JCI147836
- Blackstone, C.; Prinz,Keeping in shape, W.A.. Elife 2016, 5, e20468 . DOI: https://doi.org/10.7554/eLife.20468
- Perkins, H.T.; Allan, V.J.; Waigh, T.A. Network organisation and the dynamics of tubules in the endoplasmic reticulum. Sci. Rep. 2021, 11, 16230. DOI: https://doi.org/10.1038/s41598-021-94901-2
- Hofmann, C.; Katus, H.A.; Doroudgar, S. Protein Misfolding in Cardiac Disease. Circulation 2019, 139, 2085–2088. DOI: https://doi.org/10.1161/CIRCULATIONAHA.118.037417
- Jin, J.K.; Blackwood, E.A.; Azizi, K.; et al. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signalling Pathways in the Heart. Circ. Res. 2017, 120, 862–875. DOI: https://doi.org/10.1161/CIRCRESAHA.116.310266
- Bhatta, M.; Chatpar, K.; Hu, Z.; et al. Reduction of Endoplasmic Reticulum Stress Improves Angiogenic Progenitor Cell function in a Mouse Model of Type 1 Diabetes. Cell Death Dis. 2018, 9, 467. DOI: https://doi.org/10.1038/s41419-018-0501-5
- Yao, Y.; Lu, Q.; Hu, Z.; et al. A non-canonical pathway regulates ER stress signalling and blocks ER stress-induced apoptosis and heart failure. Nat. Commun. 2017, 8, 133. DOI: https://doi.org/10.1038/s41467-017-00171-w
- Binder, P.; Wang, S.; Radu, M.; et al. Pak2 as a Novel Therapeutic Target for Cardioprotective Endoplasmic Reticulum Stress Response. Circ. Res. 2019, 124, 696–711. DOI: https://doi.org/10.1161/CIRCRESAHA.118.312829
- Al-Yacoub, N.; Colak, D.; Mahmoud, S.A.; et al. Mutation in FBXO32 causes dilated cardiomyopathy through up-regulation of ER-stress mediated apoptosis. Commun. Biol. 2021, 4, 884. DOI: https://doi.org/10.1038/s42003-021-02391-9
- Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. DOI: https://doi.org/10.1038/nrm3270
- Liu, W.; Zi, M.; Chi, H.; et al. Deprivation of MKK7 in cardiomyocytes provokes heart failure in mice when exposed to pressure overload. J. Mol. Cell Cardiol. 2011, 50, 702–711. DOI: https://doi.org/10.1016/j.yjmcc.2011.01.013
- Chowdhury, S.K.; Liu, W.; Zi, M.; et al. Stress-Activated Kinase Mitogen-Activated Kinase Kinase-7 Governs Epigenetics of Cardiac Repolarization for Arrhythmia Prevention. Circulation. 2017, 135, 683–699. DOI: https://doi.org/10.1161/CIRCULATIONAHA.116.022941
- Liang, Q.; Bueno, O.F.; Wilkins, B.J.; et al. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signalling. EMBO J., 2003, 22, 5079–5089. DOI: https://doi.org/10.1093/emboj/cdg474
- Shao, Z.; Bhattacharya, K.; Hsich, E.; et al. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ. Res. 2006, 98, 111–118. DOI: https://doi.org/10.1161/01.RES.0000197781.20524.b9
- Petrich, B.G.; Eloff, B.C.; Lerner, D.L.; et al. Targeted activation of c-Jun N-terminal kinase in vivo induces restrictive cardiomyopathy and conduction defects. J. Biol. Chem., 2004, 279, 15330–15338. DOI: https://doi.org/10.1074/jbc.M314142200
- Wang, J.; Wang, H.; Chen, J.; et al. GADD45B inhibits MKK7-induced cardiac hypertrophy and the polymorphisms of GADD45B is associated with inter-ventricular septum hypertrophy. Biochem. Biophys. Res. Commun. 2008, 372, 623–628. DOI: https://doi.org/10.1016/j.bbrc.2008.05.122
- Pandey, V.K.; Mathur, A.; Kakkar, P. Emerging role of Unfolded Protein Response (UPR) mediated proteotoxic apoptosis in diabetes. Life Sci. 2019, 216, 246–258. DOI: https://doi.org/10.1016/j.lfs.2018.11.041
- Liu, C.L.; Li, X.; Gan, L.; et al. High-content screening identifies inhibitors of the nuclear translocation of ATF6. Int. J. Mol. Med. 2016, 37, 407–414. DOI: https://doi.org/10.3892/ijmm.2015.2442
- Gao, G.; Xie, A.; Zhang, J.; et al. Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ. Arrhythm. Electrophysiol. 2013, 6, 1018–1024. DOI: https://doi.org/10.1161/CIRCEP.113.000274
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; et al. Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signalling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Front. Pharmacol. 2019, 10, 977. DOI: https://doi.org/10.3389/fphar.2019.00977
- Dorsch, L.M.; Schuldt, M.; dos Remedios, C.G.; et al. Protein Quality Control Activation and Microtubule Remodelling in Hypertrophic Cardiomyopathy. Cells 2019, 8, 741. DOI: https://doi.org/10.3390/cells8070741
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; et al. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. DOI: https://doi.org/10.1161/CIRCRESAHA.116.307474
- Wang, X.; Deng, Y.; Zhang, G.; et al. Spliced X-box Binding Protein 1 Stimulates Adaptive Growth Through Activation of mTOR. Circulation 2019, 140, 566–579. DOI: https://doi.org/10.1161/CIRCULATIONAHA.118.038924
- Duan, Q.; Ni, L.; Wang, P.; et al. Deregulation of XBP1 expression contributes to myocardial vascular endothelial growth factor-A expression and angiogenesis during cardiac hypertrophy in vivo. Aging Cell 2016, 15, 625–633. DOI: https://doi.org/10.1111/acel.12460
- Steiger, D.; Yokota, T.; Li, J.; et al. The serine/threonine-protein kinase/endoribonuclease IRE1α protects the heart against pressure overload-induced heart failure. J. Biol. Chem. 2018, 293, 9652–9661 DOI: https://doi.org/10.1074/jbc.RA118.003448
- Hu, H.; Tian, M.; Ding, C.; et al. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083. DOI: https://doi.org/10.3389/fimmu.2018.03083
- Fu, H.Y.; Okada, K.; Liao, Y.; et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361–369 DOI: https://doi.org/10.1161/CIRCULATIONAHA.109.917914
- Finnson, K.W.; Almadani, Y.; Philip, A. Non-canonical (non-SMAD 2/ 3) TGF-β signalling in fibrosis: Mechanisms and targets. Semin. Cell Dev. Biol. 2020, 101, 115–122. DOI: https://doi.org/10.1016/j.semcdb.2019.11.013
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; et al. Cardiac fibrosis: new insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. DOI: https://doi.org/10.7150/ijbs.28103
- Leivonen, S.K.; Lazaridis, K.; Decock, J.; et al. TGF-β-elicited induction of tissue inhibitor of metalloproteinases (TIMP)-3 expression in fibroblasts involves complex interplay between Smad3, p38α, and ERK1/2. PLoS One 2013, 8, e57474 . DOI: https://doi.org/10.1371/journal.pone.0057474
- Sadoshima, J.; Montagne, O.; Wang, Q.; et al. The MEKK1-JNK pathway plays a protective role in pressure overload but does not mediate cardiac hypertrophy. J. Clin. Invest. 2002, 110, 271–279. DOI: https://doi.org/10.1172/JCI0214938
- Calamaras, T.D.; Baumgartner, R.A.; Aronovitz, M.J.; et al. Mixed lineage kinase-3 prevents cardiac dysfunction and structural remodelling with pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H145–H159. DOI: https://doi.org/10.1152/ajpheart.00029.2018


