

Downloads
Download



This work is licensed under a Creative Commons Attribution 4.0 International License.
Review
Small Leucine Rich Proteoglycan in Fibrotic Diseases: New Frenemies?
Jiayu Guo 1,2, Yan Wang 1,2, Haihai Liang 1,2,3,*, and Baofeng Yang 1,2,3,*
1 Department of Pharmacology (National Key Laboratory of Frigid Zone Cardiovascular Diseases), College of Pharmacy, Harbin Medical University, Harbin 150081, China
2 Northern Translational Medicine Research and Cooperation Center, Heilongjiang Academy of Medical Sciences, Harbin Medical University, Harbin 150081, China
3 Research Unit of Noninfectious Chronic Diseases in Frigid Zone (2019RU070), Chinese Academy of Medical Sciences, Harbin 150081, China
* Correspondence: lianghaihai@ems.hrbmu.edu.cn (H.H.L); yangbf@ems.hrbmu.edu.cn (B.F.Y)
Received: 27 April 2023
Accepted: 2 June 2023
Published: 27 June 2023
Abstract: The human body is a complex organism with self-regulating ability and can cope with external pressures and challenges. To protect the body from damage during exercise or confrontations, beneath the human epidermal layer, the human body has evolved a coverall gown: the extracellular matrix (ECM). ECM provides a suitable space for the survival and activity of cells in the body, and affects the behavior of cells through signal transduction system. Proteoglycans, particularly the small leucine rich proteoglycan (SLRP) family, have been shown to be molecules that play important roles in matrix remodeling and organ fibrosis, such as by affecting ECM components or altering the intracellular environment. But in recent years reports of SLRP families, their manifestations in different organs have not been consistent. Recent studies suggest that proteoglycans entering the blood in a soluble form hold promise as diagnostic biomarkers of organ fibrosis and may provide novel therapeutic strategies for fibrotic diseases. Herein, we discuss and review studies of SLRPs in multi-organ fibrotic diseases.
Keywords:
small leucine rich proteoglycan organ fibrosis extracellular matrix1. Introduction
Fibrosis can occur in multiple organs, and the main pathological changes are increased fibrous connective tissue and decreased parenchymal cells within the organ tissues. The continuous progress can cause the destruction and reduction of organ structure and even failure, seriously threatening human health and life. Pathological conditions in organs such as the heart, liver, lungs and kidneys have been implicated in this process [1]. Besides that, the effect of environment on organ fibrosis is also critical. Epidemiological evidence indicates that emergency admissions and mortality are significantly higher in frigid zone, especially for adverse cardiovascular events. Both clinical and experimental studies have revealed that cold stress triggers a variety of pathological and pathophysiological insults, including ventricular wall thickening and myocardial interstitial fibrosis [2]. During development, fibroblasts are architects of the extracellular matrix (ECM) and tissue architecture, tissues and organs often become relatively quiet as they reach maturity. Following tissue injury, they are rapidly activated and are characterized by proliferation, migration, increased ECM cross-linking, and physical contraction [3]. ECM acts as a binder, holding all the cells of the tissue in place. It also forms some specialized structures that not only function as physical scaffolds but also regulate many cellular processes including growth, migration, differentiation, homeostasis and morphogenesis [4,5].The components and assembled forms of the ECM are determined by the cells produced and are tailored to the specialized functional needs of the tissue. At the general level, ECM has two main components, namely fibers and matrix [6,7].The fibrous proteins include collagen and elastin, and the matrix mainly includes fibronectin, proteoglycans, and laminin, several receptors connect the intracellular and extracellular signaling [8] (Figure 1).
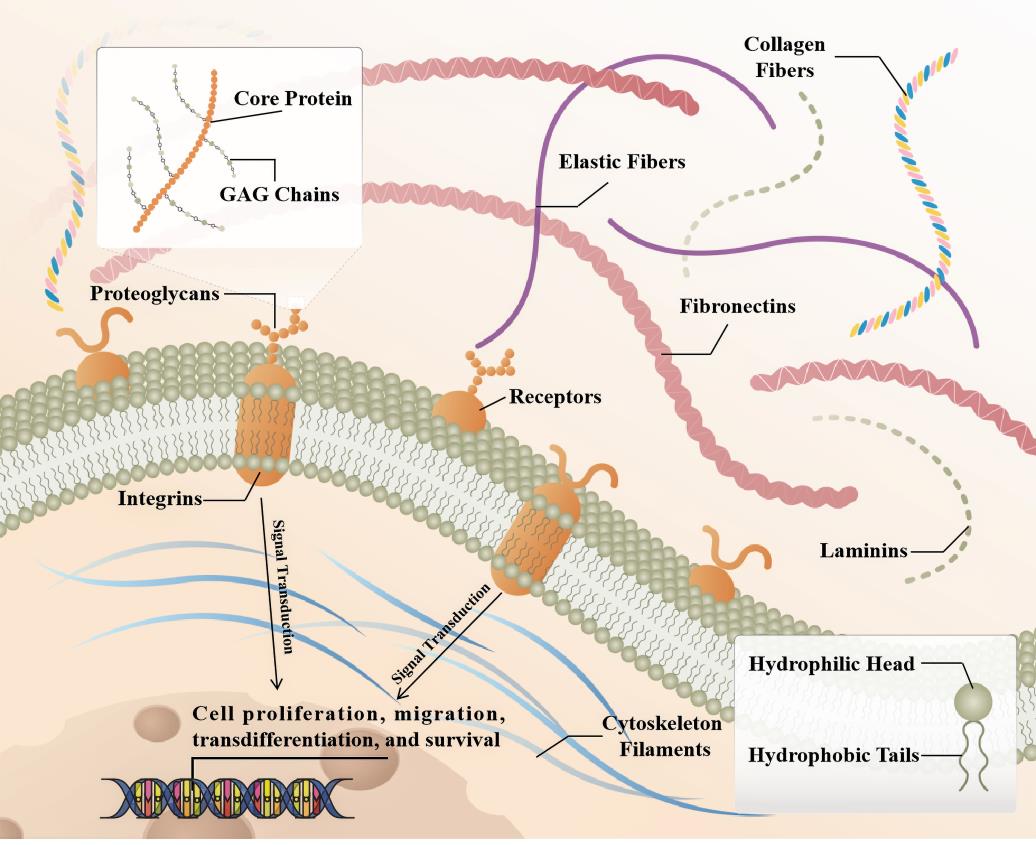
Figure 1. Schematic representation of the SLRP structure in fibrotic organ's extracellular matrix (ECM). Briefly, collagen fibrils provide tensile strength to the ECM, limiting the expansion of the tissue. In addition to playing a role in ECM assembly, laminin, elastin, and fibronectin also participate in ECM cell interactions by acting as ligands for cell surface receptors, such as integrins. SLRPs consist of a core protein with covalently attached GAG chains, distributed in collagen fibrils, able to bind to extracellular cytokines or to receptors on the cell membrane, affecting intracellular signaling pathways that in turn regulate cell fate.
Proteoglycans are glycosylated proteins. They consist of a core protein with covalently attached glycosaminoglycan (GAG) chains that largely determine proteoglycan function [9]. GAGs are among the most negatively charged molecules in mammalian tissues, allowing reversible and irreversible interactions with positively charged partners, including matrix proteins, cytokines, chemokines, pathogens, growth factors, and proteases [10]. The SLRP family consists of a group of low molecular weight secretory proteoglycans, named after their leucine-rich repeats (LRRs). Precollagen self-assembles to form triple helix collagen molecules and combines to form collagen fibers. This process is regulated by auxiliary ECM molecules, including SLRP family [11]. SLRPs bind collagen fibrils and affect collagen fibrillogenesis by regulating collagen fibril diameter and interfibrillar spacing in the ECM. SLRP family members that have been studied in organ fibrosis include Lumican (LUM), Biglycan (BGN), Decorin (DCN), Asporin (ASPN) and Osteoglycan (OGN), among others [10].The SLRP family consists of 18 different proteoglycans characterized by a relatively small molecular weight protein core of 36-42 kDa and the presence of LRRs in the structure. During synthesis, SLRPs are glycosylated and posttranslationally modified in the Golgi apparatus and secreted directly into the extracellular milieu by exocytosis [12].When bound to the ECM, SLRPs interact with various types of collagens and regulate fibrillar growth and organization, cell matrix interactions, ECM assembly and tissue function. In addition, in the ECM, SLRPs can exist either in a matrix bound, sequestered form or in a soluble form. Matrix bound SLRPs are able to sequester signaling mediators in the ECM, thereby interfering with different signaling cascades [13,14].SLRPs are classified into five classes based on evolutionary conservation, protein and genome homology, and chromosomal organization [9].The class I SLRP DCN, BGN and ASPN, class II LUM and FMOD, and class III OGN are the best characterized members of the SLRP family and have been extensively studied in fibrotic diseases in recent years. Targeting fibrosis to treat disease remains challenging, and further elucidation of the cellular and molecular mechanisms of fibrosis is required to develop therapies that can translate into positive clinical outcomes for patients.
2. The Expression of SLRP Family in Organ Fibrosis
Class I: As early as 1993, the author found that the mRNA of BGN in lung tissue increased after bleomycin perfusion, while the mRNA of DCN decreased on the 10th day, reaching 20% of the control group [15]. The ECM of the human atrial appendage was analyzed by proteomics, and DCN expression was increased in patients with atrial fibrillation [16]. The relative abundance of DCN and BGN strongly increased during liver fibrosis [17]. Experimental animal models of unilateral ureteral ligation, resulted in overexpression of DCN, BGN, Fibrin-1 and Fibrin-2 [18]. DCN and BGN proteins were not detected in normal glomeruli. DCN accumulated in amyloid deposits and both DCN and BGN were found in glomeruli in areas of fibrous tissue in the urinary lumen and in areas of tubulointerstitial fibrosis [19]. BGN expression was significantly increased in patients and mice with myocardial infarction [20]. After TAC, the expression of BGN in fibroblasts was up-regulated, but the expression of BGN in cardiomyocytes, endothelial cells or leukocytes was not up-regulated [21]. Hepatic BGN expression is significantly increased in fibrotic patients and mouse models of liver fibrosis. BGN promotes liver fibrosis by positively regulating HSP47 to regulate ECM deposition and hepatic stellate cells (HSC) activation [22]. In other organs, DCN was downregulated while BGN was upregulated during bladder fibrosis [23]. ASPN expression was increased after pressure overload or cardiac ischemia/reperfusion injury [24]. Meanwhile, ASPN expression was upregulated in the lungs of patients with IPF and mouse models of pulmonary fibrosis and was predominantly localized to α-SMA+ myofibroblasts [25]. However, the expression and role of ASPN in renal fibrosis and liver fibrosis have not been clearly reported. ECM2 was detected to be upregulated in patients with IPF compared to healthy controls at mRNA levels [26].
Class II: LUM was increased in experimental and clinical heart failure [27], and likewise, LUM, an ECM proteoglycan, was also upregulated in familial hypertrophic cardiomyopathy (HCM) and colocalized with fibrillar collagen throughout the fibrotic area in HCM [28]. Moderate absence of LUM in mice attenuated cardiac fibrosis and ameliorated diastolic dysfunction after pressure overload [29]. In lung tissue from patients with pulmonary fibrosis with relatively normal lung function, LUM was present at low levels throughout the tissue, whereas patients with advanced disease had significant LUM expression in fibrotic lesions and was particularly prominent in areas adjacent to the epithelial layer [30]. LUM was upregulated in the kidney and liver, respectively, in the unilateral ureteral obstruction (UUO) model and CCl4 induced liver fibrosis [31,32], and expression was also increased in the fibrotic joint capsule [33]. Cardiac fibromodulin (FMOD) was upregulated 3-10 fold in heart failure patients and mice. Both cardiomyocytes and cardiac fibroblasts expressed FMOD and its expression increased upon pro-inflammatory stimuli. After aortic banding, FMOD-/- mice developed mildly aggravated hypertrophic remodeling in the left ventricle with increased cardiomyocyte size and altered leukocyte infiltration compared to wild-type mice [34]. Hepcidin messenger RNA and protein levels were higher in liver samples from patients with cirrhosis than in controls. Recombinant FMOD promoted the proliferation, migration and invasion of haematopoietic stem cells and promoted their fibroblastic activity, leading to liver fibrosis in mice [35]. Immunocytochemistry results showed strong immunostaining for FMOD in the injured area of rat lung tissue at 14 and 28 d after BLM administration [36], but in the study by G Westergren-Thorsson et al., in BLM induced pulmonary fibrosis the message for FMOD remained unchanged throughout the study period [15]. In addition, FMOD was expressed at higher levels in chronic pancreatitis (CP) fibrotic tissues [37].
Class III: OGN, a proteoglycan released from bone and muscle, is associated with markers of metabolic health. Higher OGN was associated with higher VO2 peak and a higher circulating glucose level [38]. OGN expression levels were higher in mouse infarcted left ventricle and ischemic heart disease patient hearts, as well as in lung tissues of ILD mice [39] (Figure 2).
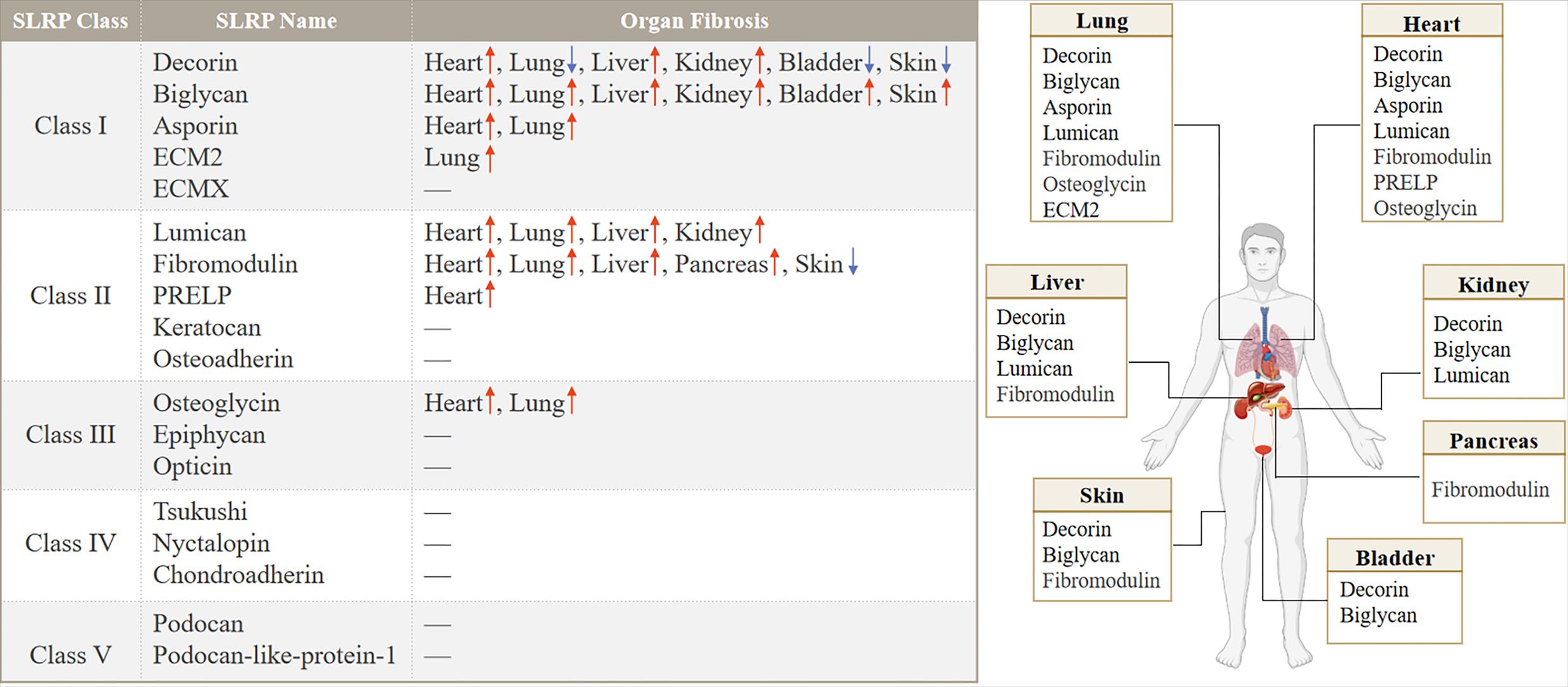
Figure 2. Distribution and expression of SLRP family members in organ fibrosis.
3. Functional Diversity of SLRP Family Members in Multi-organ Fibrosis
3.1. Cardiac Fibrosis
Fibrosis is a common end-stage feature of virtually every heart disease, centered on cardiomyocyte hypertrophy with excessive deposition of ECM. Over time, this accumulation leads to impaired cardiac systolic and diastolic function and an increased propensity for arrhythmias and death. In the adult heart, fibroblasts continuously alter the microenvironment by degrading and depositing ECM [40]. In many cases, cardiac fibrosis is reparative, whereas the adult mammalian heart has negligible regenerative capacity and it can only heal by forming a scar, reflecting replacement of dead cardiomyocytes by a collagen scar that initially helps stabilize the heart, enhances tissue integrity, but extensive fibrosis can impair cardiac function [40]. Sudden death of a large number of cardiomyocytes stimulates an inflammatory response with subsequent activation of reparative myofibroblasts, leading to scar formation. The scar lacks contractile capacity but plays a key protective role, maintaining the structural integrity of the chamber and, in the case of transmural infarction, preventing catastrophic mechanical complications such as cardiac rupture [41] (Figure 3).
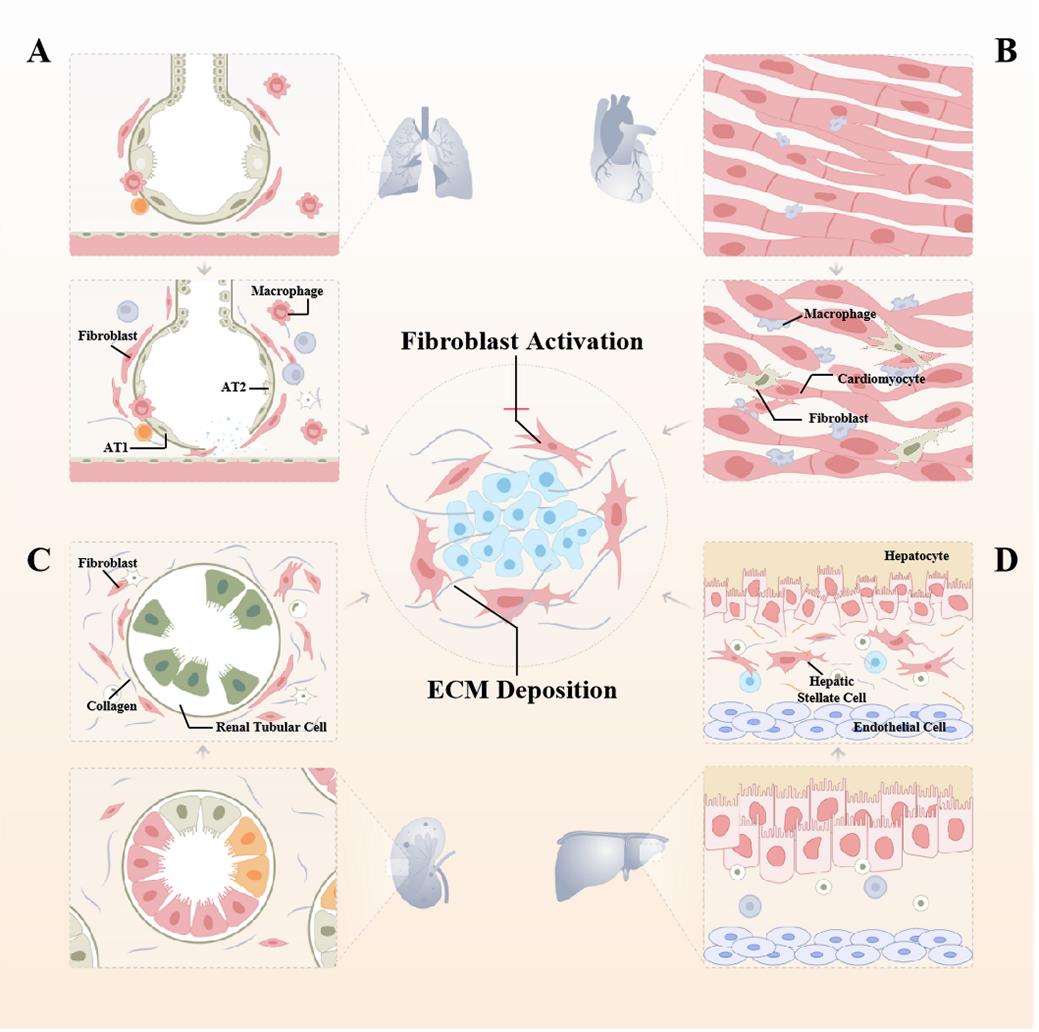
Figure 3. The central role of fibroblast activation in the pathogenesis of various organ fibrosis. (A) Pulmonary fibrosis is a common alveolar disease in which external stimuli repeatedly damage alveolar epithelial cells, and healthy lung tissue is gradually replaced by excessive amounts of interstitial cells and ECM, leading to the destruction of the alveolar structure, progressive densification of alveolar areas, and ultimately respiratory failure and death. (B) When myocardial fibrosis occurs, because of cardiomyocyte hypertrophy and ventricular remodeling, the normal structure of the myocardium is destroyed, the patient's myocardial elastic ability is impaired, and the normal contraction and diastole cannot be performed, and the ejection function of the heart cannot meet the needs of myocardial tissue. Pre-existing cardiomyocytes interdigitate and are arranged in a disorganized manner with a massive proliferation of fibrotic tissue, and the heart is progressively calcified and stiffened. (C) Nephron loss can trigger wound repair to further progress to interstitial fibrosis. Renal tubular epithelial cells undergo partial EMT and exhibit changes such as senescence and apoptosis that ultimately lead to tubular atrophy, promoting proinflammatory and profibrotic mediator secretion. (D) Regeneration after repeated destruction of hepatocytes, diffuse excessive deposition, and abnormal distribution of ECM such as collagens, glycoproteins, and proteoglycans in the liver, which contribute to the pathological repair response of the liver to chronic injury, is a key step in the progression of various chronic liver diseases to cirrhosis. As the main cell population in the liver synthesizing ECM, HSCs activation, proliferation, and transformation are important processes in liver fibrosis.
3.1.1. Decorin
In the heart, DCN deficiency did not alter collagen concentration or cross-linking but resulted in uneven collagen fibre diameter and lateral wall filling within the infarct scar [42]. It is well known that the collagen superfamily plays a dominant role in maintaining the integrity of various tissues, and the onset of fibrosis is characterized by extracellular collagen deposition, but the lack of collagen in the early stage may lead to impaired repair. Sara M Weis et al. found that two weeks after acute MI, DCN-/- mice had increased infarct size, enhanced ventricular remodelling and reduced ventricular function [42]. This may be attributed to the fact that abnormal collagen fibrils adversely affect postinfarction mechanics and ventricular remodeling. In addition, the proteoglycan DCN was a known TGF-β1 inhibitor [43]. The mRNA and protein levels of DCN were increased in patients with heart failure treated with mechanical circulatory support (MCS), and DCN molecules may be involved in reverse cardiac remodeling by directly inhibiting the profibrotic effects of the TGF-β/Smad2 pathway [44]. Furthermore, DCN gene delivery regulated TGF-β/Smad and p38 MAPK signaling pathways inhibited myocardial fibrosis in spontaneously hypertensive rats [45]. Several ECM derived proteoglycans and proteins, including DCN, BGN, and Endostatin have been identified as strong inducers of autophagy. In contrast, Laminin α2, Perlecan, and LUM exerted opposite effects by inhibiting autophagy [46]. Specifically, DCN enhanced Beclin-1 expression and induced phosphorylation of AMPK, which in turn stimulated Peg3 dependent autophagy in endothelial cells by interacting with vascular endothelial growth factor receptor (VEGFR2) [47,48]. In addition, DCN regulated autophagy and mitophagy through direct interaction with members of the subfamily of cell surface receptor tyrosine kinases (RTKs) in endothelial cells [49].
3.1.2. Biglycan
BGN deficiency led to spontaneous aortic dissection and rupture in mice, 50% of Biglycan deficient male mice died suddenly within the first 3 months of birth, and autopsy revealed massive hemorrhage in the thoracic or abdominal cavity, with histology showing aortic rupture. Transmission electron microscopy and biomechanical testing revealed abnormal collagen fibril structure and reduced tensile strength in the aorta of Biglycan-/- mice [50]. BGN knockout mice had increased mortality after myocardial infarction due to frequent left ventricular (LV) rupture, which manifested as aggravated LV dilation and severely impaired LV function. Tensile strength of LV was reduced and collagen matrix organization was severely impaired in BGN knockout mice 21 days after MI [20], indicating that BGN deficiency is essential for the structural and functional integrity of the aortic wall and postinfarction heart. BGN participated in pathological cardiac remodeling by binding to TGF-β [51]. Phenotypic characterization of BGN deficient fibroblasts revealed increased proliferation, and the expression of α-SMA and TGF-β receptor II and phosphorylation of Smad2 were markedly elevated after myocardial infarction in BGN(-/0) mice, indicating that BGN deficiency promoted myofibroblast differentiation and proliferation probably in response to increased TGF-β and Smad2 signaling [52]. In contrast to the above results, after 9 weeks of long-term TAC, the ablation of BGN attenuated the development of myocardial hypertrophy and fibrosis. In vitro, BGN induced cardiomyocyte hypertrophy in neonatal rats, and led to the activation of hypertrophy gene program and the expression of hypertrophy related genes Rcan1, Abra and Tnfrsf12a [21]. The combination of BGN overexpression and Ang II infusion resulted in a significant increase in coronary artery VSMC proliferation and migration, as well as increased perivascular fibrosis [20]. Inconsistencies in the results of these studies may be due to different models, and thus different functions and animal phenotypes of BGN in acute MI and chronic cardiac fibrosis.
3.1.3. Lumican
LUM was an inflammation-related ECM-targeting proteoglycan that is known to bind to collagen. The production and release of LUM by cardiac fibroblasts was induced by mechanical and proinflammatory stimuli, and LUM may induce collagen cross-linking by increasing the production of type I collagen and lysine oxidase in cardiac fibroblasts [27]. LUM-KO mice had increased mortality 1-14 days after aortic banding (AB). Echocardiography showed increased left ventricular dilation, altered hypertrophic remodeling, and aggravated systolic dysfunction in the surviving LUM-KO 1-10 week post AB. LUM-KO hearts showed a reduction in collagen expression and AB post cross linking. Lack of LUM attenuated collagen cross-linking in pressure overloaded hearts, leading to increased mortality, dilation, and systolic dysfunction in mice [53]. In other experimental models, LUM-/- mice were prone to aging and isoproterenol induced myocardial fibrosis. After isoproterenol induced cardiac fibrosis, LUM-/- mice had reduced blood pressure and impaired cardiac function, and exhibited severe cardiac fibrosis and disorganized cardiomyocytes [54]. But in a recent study, Chloe Rixon et al. suggested that LUM may promote the formation of thicker collagen fibers in HCM [28]. At the cellular level, Kristin V T Engebretsen et al. showed that LUM increased TGF-β production and phosphorylation of Smad3 in cardiac fibroblasts to stimulate cardiac fibrosis [27]. This may seem somewhat paradoxical, as it is generally believed that activation of TGF-β signaling pathways stimulated fibroblast activation and then aggravated the fibrotic process. However, this may be due to the fact that LUM is beneficial for the early repair of injury, so overexpression LUM transgenic mice need to be constructed to examine its role in cardiac fibrosis.
3.1.4. Fibromodulin
FMOD bound to specific portions of the collagen domain and forms a complex with lysyl oxidase to direct the enzyme to specific sites of cross-linking [55]. In vitro, overexpression of FMOD decreased the expression of LOX and TGM2 in cardiac fibroblasts. Furthermore, the reduced expression of the ECM protein Periostin suggested a direct effect of FMOD on the profibrotic properties of cardiac fibroblasts, but collagen quantity and cross-linking are comparable between FMOD-/- and wild-type mice [34].
3.1.5. Osteoglycin
OGN promoted cross-linking of collagen fibers and was essential for proper collagen assembly in the diseased heart. Increased expression of OGN in the infarct scar promoted collagen maturation after myocardial infarction and prevented cardiac destruction and adverse remodeling [39]. However, OGN was negatively correlated with the expression of collagen in the study of Sophie deckx et al. OGN deficient cardiac fibroblasts were more susceptible to premature senescence, with a trend toward increased senescence associated p16 expression [56]. OGN deficiency caused diastolic dysfunction in aged mice. OGN limited the expression of proinflammatory molecules IL-1β, ICAM-1, and MCP-1 by macrophages and reduced cardiac fibroblast proliferation and TGF-β-mediated collagen production [57]. OGN-/- mice exhibited enhanced myocardial interstitial fibrosis and significantly more severe cardiac dysfunction after Ang II infusion, but OGN deficiency did not affect cardiomyocyte hypertrophy nor alter blood pressure. Mechanistically, OGN negatively regulated cardiac fibrotic remodeling by inhibiting cardiac myofibroblasts proliferation and migration through LPA3 mediated and Rho/ROCK dependent inhibition of MT1-MMP translocation, MMP2 activation, and EGFR transactivation [58]. However, the role of OGN was bifacial. In a myocarditis mouse model, OGN silencing had an inhibitory effect on myocardial fibrosis and epithelial/endothelial mesenchymal transition. OGN gene silencing inhibited cell cycle progression while promoting apoptosis in mouse cardiac fibroblasts [59].
3.1.6. Asporin
ASPN was released by cardiac fibroblasts and attenuated TGF-β signaling [24]. However, in cardiomyocytes, ASPN promoted H9C2 cardiomyocyte apoptosis by upregulating TGF-β as well as Smad2 and Smad3 phosphorylation [60], indicating that in different cell types of the same organ, ASPN may play opposing roles. Furthermore, ASPN regulated mitochondrial bioenergetics, protecting cardiomyocytes from hypoxia mediated cell death. Utilizing ASPN derived peptides may attenuate pressure overload induced fibrosis and preserve cardiac function [24].
3.1.7. Proline and Arginine Rich End Leucine Rich Repeat Protein (PRELP)
Overexpression of PRELP increased infarct size and interstitial fibrosis area, and PRELP promoted myocardial fibrosis and ventricular remodeling after acute myocardial infarction through Wnt/β-catenin signaling pathway [61].
3.2. Liver Fibrosis
Liver fibrosis is a dynamic process responsible for driving excessive accumulation of ECM components, maintained by heterogeneous hepatic myofibroblasts [62]. Cirrhosis is characterized by structural changes, including the formation of parenchymal regenerative nodules surrounded by fibrotic septa, and significant alterations in the vascular architecture of organs, which in turn can lead to the development of portal hypertension and associated complications (e.g., variceal hemorrhage, hepatic encephalopathy, ascites). Chronic hepatocyte injury leads to the release of damage associated patterns (DAMPs) and apoptotic bodies that activate hepatic stellate cells (HSCs) and recruit immune cells. Complex multidirectional interactions between activated HSCs and Kupffer cells as well as innate immune cells promote transdifferentiation into proliferating and ECM-producing myofibroblasts [62]. HSCs in the normal liver reside in the subendothelial space of Disse and establish close contacts with surrounding hepatocytes and nerve endings through their cytoplasmic processes. Activated hepatic stellate cells, portal fibroblasts and bone marrow-derived myofibroblasts have been identified as the major collagen producing cells in the injured liver through their activation and transdifferentiation into MF like cells under conditions of chronic liver injury. Emerging antifibrotic treatments aim to inhibit the accumulation of fibroblasts and/or prevent the deposition of ECM proteins [63].
3.2.1. Decorin and Biglycan
DCN-/- mice not only had more severe liver fibrosis induced by experiment, but also had significantly delayed the healing process compared with wild-type mice. The excessive deposition of connective tissue in the liver of DCN-/- mice was due to increased production rather than degradation damage [64]. Exogenous protein DCN could attenuate CCl4 induced liver fibrosis in mice and promote liver regeneration after partial hepatectomy in fibrotic mice. In the experimental group, the degree of fibrosis was reduced, as was the collagen fiber content, TGF-β and α-SMA expression. Treatment with bone marrow-derived mesenchymal stem cells (BM-MSCs) in combination with DCN protein resulted in strong protection against liver fibrosis by inhibiting TGF-β/Smad signaling [65]. DCN had a protective role in liver fibrogenesis as its genetic ablation in mice led to enhanced matrix deposition, impaired matrix degradation, and activation of hepatic stellate cells [66]. But Kornélia Baghy et al. found that two major axes of TGF-β-induced signaling pathways were affected in DCN -/- mice, namely ERK1/2 and Smad3 were activated, while no significant difference was observed in phosphorylated Smad2 [64]. Cells dying by ferroptosis released DCN, which then acted as an alarming signal to trigger innate and adaptive immune responses. Extracellular DCN bound to its receptor advanced glycation end product specific receptor (AGER) on macrophages to trigger proinflammatory cytokine production in an NF-κB dependent manner [67]. In response to tissue injury, the ECM may become saturated and unable to sequester excess biglycan. BGN was released from the ECM as a danger signal, and at the same time proteases can cleave the ECM bound BGN to release soluble BGN, which interacted with innate immune receptors toll like receptors 2 (TLR2) and 4 (TLR4) to initiate a sustained inflammatory response [68]. Meanwhile, by interacting with TLR4, BGN could mediate the downstream effects of pro-inflammatory and pro-autophagy, depending on the choice of receptors CD14 and CD44, respectively [68]. BGN promoted the enhancement of Beclin-1 via innate immune TLR-4 and its co-receptor CD44, triggered LC3-II conversion and p62 recruitment to promote autophagic flux in macrophages, but not in endothelial cells [68-70]. In addition, BGN evoked autophagy further contributes to anti-inflammatory M2 macrophage polarization, inflammation resolution, and tissue repair [68].
3.2.2. Lumican
In liver fibrosis model, LUM null could not protect against liver injury or inflammation, but had a protective effect on fibrotic progression, which seemed to be downstream of collagen production and mediated by the combined effects of impaired collagen fibrogenesis, increased matrix turnover and enhanced proliferative response [31]. LUM significantly decreased AMPK activity and inhibited autophagy. The combination of LUM and Gemcitabine increased mitochondrial damage, reactive oxygen species (ROS) production and cytochrome-C release, suggesting lumican induced disruption of mitochondrial function [71].
3.3. Pulmonary Fibrosis
In patients with pulmonary fibrosis, healthy tissue is replaced by altered ECM and alveolar architecture is disrupted, leading to decreased lung compliance, disrupted gas exchange, and ultimately respiratory failure and death. Pulmonary fibrosis is currently thought to originate from abnormal repair following repeated injury to the alveolar epithelium [72]. Whereas pulmonary fibrosis is mainly characterized by excessive proliferative activation of stimulated fibroblasts, differentiation into myofibroblasts, secretion of large amounts of collagen and matrix proteins, increased migratory capacity, expression of α-SMA and formation of contractile bundles, alterations in mechanical forces and structural microenvironment and increases in certain factors such as TGF-β can further promote fibroblast activation, leading to the formation of a fibrous scar with honeycomb cysts, ultimately resulting in disrupted lung architecture and loss of function [73,74].
3.3.1. Decorin and Biglycan
In vitro, the cell supernatant of AdDec-infected cells eliminated the biological activity of TGF-β in a dose-dependent manner. Single administration of AdDec in vivo was beneficial to produce local lung environment, and effectively blocked the fibrosis response to bleomycin by inhibiting TGF-β [75]. DCN and BGN were able to bind and inhibit TGF-β activity in vitro [22,76]. However, in vivo, overexpression of DCN significantly attenuated TGF-β-induced lung fibrosis, whereas BGN had no effect [76]. Soluble DCN acted as a signaling molecule for the IGF-IR, thereby protecting epithelial cells from apoptosis or inducing fibrillin-1 production by fibroblasts, and Allawadhi P et al. suggested that DCN could be a strategy to improve COVID-19 [77]. However, DCN exerted anti-fibrotic effects to some extent, as DCN also induced IL-10 anti-inflammatory gene expression through TLR2/4 [78]. It has been reported that DCN may play a negative regulatory role in the formation of central fibrosis in lung adenocarcinoma [79].
3.3.2. Lumican
LUM levels in BALF were significantly higher in acute respiratory distress syndrome (ARDS) patients than in ventilated or spontaneously breathing controls, and Lumican increased α-SMA, COL1A1, and COL3A1 expression in primary human lung fibroblasts [80]. Secretion of inflammatory signals by fibroblasts TNF-α stimulated secretion of the small leucine rich proteoglycan LUM, which further promoted human fibroblast differentiation. And that LUM induced fiber cell differentiation was based on α2β1,αMβ2 and αXβ2 integrin functioning [30]. An increase in ERK phosphorylation and Slug was found in both LUM treated lung fibroblasts and small airway epithelial cells (SAECS). LUM triggered the transdifferentiation of lung fibroblasts into myofibroblasts, and the epithelial mesenchymal transition of SAECS, possibly through the ERK/Slug pathway [80].
3.3.3. Others
In lung tissue, the function of OGN was closely related to the Wnt signaling pathway, and results from Songtao Shi et al. showed that overexpression of microRNA-140 reduced proliferation and lung fibrosis by leading to apoptosis of lung fibroblasts and increased Wnt3a expression via downregulation of OGN [81]. In a pulmonary fibrosis model, FMOD deficient mice were not protected, but the composition of the matrix was affected and FMOD deficient mice had reduced type I collagen. Also BGN was increased while DCN was decreased in animals with FMOD deficiency [82]. ASPN reduction in lung fibroblasts inhibits the TGF-β/Smad signaling pathway and myofibroblast differentiation by regulating the stability of TGF-β receptor I (TβRI). Mechanistically, knockdown of ASPN inhibited TGF-β/Smad signaling and promoted lysosome mediated TβRI degradation by inhibiting TβRI recycling to the cell surface in an Rab11 dependent manner [25].
3.4. Kidney Fibrosis
Renal fibrosis is a common outcome in a variety of chronic kidney diseases (CKDs), mainly characterized by glomerulosclerosis and renal interstitial fibrosis, and current studies suggest that instead of being evenly distributed throughout the renal parenchyma, it starts locally, forming a distinct and clustered niche. These niches provide a unique tissue microenvironment and result in excessive deposition of ECM, leading to tissue scarring. The formation of a fibrotic niche is associated with a complex and highly dynamic series of cellular events, including tubule cell injury, inflammatory cell infiltration, myofibroblast activation, tubule atrophy and microvascular rarefaction [83]. In renal fibrosis, tubular epithelial cells are lost by cell death and the remaining cells dedifferentiate, resulting in decreased expression of characteristic epithelial markers and increased expression of mesenchymal markers [84]. In combination with environmental insults, injured epithelial cells mount aberrant responses, and glomerulosclerotic changes can promote loss of the glomerular filtration barrier and autoregulation of peritubular capillaries, ultimately leading to complete loss of glomerular filtration function, and loss of tubular to peritubular capillary interstitial transfer [85].
3.4.1. Decorin and Biglycan
Renal tissue from 18 patients with crescentic glomerulonephritis (CGN) was immunohistochemically examined, and DCN and BGN were found clustered in type I collagen rich areas, including fibrocytes and fibrous lacunae as well as interstitial fibrosis. DCN and BGN may contribute to the progression of CGNs by interacting with type I collagen in the reconstituted ECM [86], and Louise Tzung-Harn Hsieh et al. also demonstrated that soluble DCN protein played a pro-inflammatory role in unilateral ureteral obstruction [32]. Th1 and Th17 cells, the T helper (Th) subtypes, are key inducers of renal fibrosis. BGN triggered CXCL10 expression in macrophages via TLR4/TRIF dependent signaling pathway, which promoted Th1 and Th17 cell recruitment to the kidney. Meanwhile, BGN regulated renal Th17 cell infiltration by regulating CCL20 expression, leading to the development of fibrosis [87]. In addition, purified BGN stimulated macrophages to express TNF-α, iNOS, NOX2, and CCL3 via the TLR4/NF-κB pathway [88]. DCN is highly homologous with BGN, and DCN is also a high-affinity ligand of TLR2 and TLR4, which triggered the production of proinflammatory TNF-α and IL-12p70 in an NF-κB or MAPK-dependent manner [89]. DCN deletion triggered massive apoptosis of tubular epithelial cells and evoked inflammatory responses to promote renal fibrosis. However, in fibroblasts, DCN promoted IGF-IR phosphorylation and activated the Akt/PKB signaling cascade, thereby exacerbating renal fibrosis [90,91]. In renal fibroblasts, DCN bound to and induced phosphorylation of the IGF-IR, increasing Fibrillin-1 synthesis [90]. DCN has an inhibitory effect on the activity of CTGF in fibroblasts, which has been shown to have a high affinity direct interaction with CTGF through its LRR12 protein core [92].
3.5. Role of SLRPs in Other Fibrosis
DCN was found to neutralize the effects of myostatin in fibroblasts and myoblasts, while also upregulating the expression of follistatin, an antagonist of myostatin, resulting in significantly less fibrosis and better skeletal muscle regeneration [93]. DCN transfection inhibited fibroblast genes and myofibroblast formation of human corneal fibroblasts [94]. ASPN induced collagen calcification, which reduced their mechanical sensitivity and mechanical signals transmitted through the collagen network, prevented the activation and differentiation of fibroblasts into mature muscle fibroblasts, and thus effectively reconstructed the ECM [95].
Hypertrophic scar (HTS) group had significantly lower levels of DCN and FMOD and higher levels of BGN compared to normal skin [96]. In normal and over mechanically loaded porcine skin wounds, FMOD reduces scar size and increases scar tensile strength [97]. Reduced DCN, FMOD and TGF-β3 in the deeper dermis led to hypertrophic scarring [98]. In a bleomycin skin fibrosis model, FMOD-/- mice had a significant reduction in collagen fiber diameter, but the extent of skin fibrosis was similar to that in FMOD+/+ mice, so Andréasson K. et al. suggested that the impact of FMOD deficiency on the development of experimental skin fibrosis is limited [99]. FMOD-/- mice were migration inhibited by TGF-β3, and delayed dermal cell migration results in delayed wound closure and a significant increase in scar size [100]. Furthermore, Ad-FMOD induced decreased expression of TGF-β1 and TGF-β2 precursor proteins, but increased expression of TGF-β3 precursor protein and TGF-β type II receptor, which can promote wound healing, suggesting that FMOD may be a key mediator in reducing scar formation [101]. LUM promoted cutaneous wound healing by promoting the activation and contraction of wound fibroblasts via integrin α2, but not by promoting the proliferation and migration of keratinocytes [102]. Likewise, as a type II SLRP family member, PRELP potently attenuated fibroblast response to TGF-β1 stimulation and cell contractility. PRELP, as a novel natural TGF-β antagonist, had a possible dermoepidermal proadhesive ability [103]. ERK and JNK inhibitors attenuated FMOD expression. AP-1 bound to the FMOD promoter, increasing the expression of FMOD through transcriptional regulation. FMOD induced activation of pancreatic stellate cells to maintain fibrotic phenotype [37] (Figure 4).

Figure 4. Functions of SLRPs in multiple fibrosis diseases. “—” represents inhibiting fibrosis, “+” represents promoting fibrosis; “- / +” represents studies on inhibiting fibrosis and promoting fibrosis in the same organ have been reported.
4. Circulating Proteoglycan As a Diagnostic Marker
Soluble SLRPs can be generated by partial proteolytic processing of the collagen matrix or by denovo synthesis caused by stress or injury. One potential mechanism of action was that when the ECM was saturated with SLRP proteins, SLRPs became soluble to reach the extracellular fluid and subsequently entered the blood circulation [104].
To analyze the association of serum DCN levels with clinical parameters and prognosis in patients with acute exacerbation of idiopathic interstitial pneumonia (AE-IIP). Serum DCN levels were significantly lower in AE-IIP patients than in healthy subjects and patients with stable idiopathic interstitial pneumonia (SD-IIP). When all IIP patients were analyzed, serum DCN levels did not correlate with clinical parameters or prognosis. In IPF patients, serum DCN levels were significantly correlated with oxygenation, and IPF patients with low serum DCN levels had a significantly better survival than those with high serum DCN levels [105]. Development and validation of a competitive enzyme-linked immunosorbent assay (ELISA) quantifying a specific fragment of DCN degraded by cathepsin-S, DCN-CS levels were increased in lung cancer patients and IPF patients compared with healthy controls. It has potential as a novel non-invasive serum biomarker for fibrotic lung diseases [106].
Preclinical animal models and human studies have recently identified soluble BGN as a key initiator and modulator of various inflammatory kidney diseases [32]. One study suggested that measurement of BGN levels in serum may aid in the diagnosis of heart failure patients who may benefit from lipid-lowering therapy with statins [107]. Serum BGN was associated with liver fibrosis and inflammation, and BGN was increased in the significant fibrosis group compared to the mild fibrosis group. BGN was an independent predictor of significant fibrosis in multivariate analysis [108]. Consistently, serum BGN levels were significantly higher in patients with chronic hepatitis B than in healthy controls. There was a significant positive correlation between serum BGN levels and fibrosis stage. In addition, serum BGN levels were positively correlated with necroinflammatory activity with statistical significance [109].
Multiple studies have found that OGN may be a biomarker for a variety of diseases, and OGN was identified as a possible biomarker in amniotic fluid in a study that identified women at risk for preterm labor and delivery [110]. OGN holds promise as a biomarker for vascular diseases. At 1-year follow-up of patients who underwent coronary angiography for acute coronary syndrome or stable angina, circulating OGN levels were associated with major adverse cardiovascular events [111]. ECM remodeling plays a key role in the progression and complications of fibrosis in multiple organs. Five biomarkers related to remodelling biology were assessed in the diagnostic follow-up of HF patients, of which OGN was most associated with events [112]. Compared with patients with nonischemic HF, those with a history of myocardial infarction had increased circulating OGN levels. OGN levels were negatively correlated with ventricular volumes and correlated with fibrosis, suggesting adverse remodeling and poor prognosis [39,113]. In a multivariate analysis of 383 patients with chronic kidney disease stages 3 to 4, lower levels of proteinuria and hemoglobin and higher levels of C-reactive protein were significantly associated with higher levels of OGN. In the non-diabetic group, each 1 ng/ml increase in serum OGN was associated with all-cause mortality and the composite outcome. However, in the diabetic group, OGN levels were not associated with mortality, major adverse cardiovascular and cerebrovascular events, or the composite outcome [114].
Serum soluble LUM levels were used as a potential prognostic factor for predicting poor outcomes after aortic surgery [115]. Multislice computed tomographic angiography (MSCTA) was used to determine the severity of acute aortic dissection (AAD). Serum was collected from patients on admission, and LUM levels were measured by enzyme-linked immunosorbent assay. LUM levels were significantly higher in AAD patients compared to healthy volunteers. LUM may be a potential marker to aid in the diagnosis and screening of AAD and can be used to predict the severity of AAD [116]. Linear regression analysis revealed a significant correlation between hepatic collagen fractional synthesis rate (FSR) and plasma LUM FSR in all subjects, as well as in subjects with NASH+ fibrosis only. Turnover of plasma fluorescent protein was approximately 3-fold faster than that of liver collagen. Similar to hepatic collagen FSR, plasma LUM FSR was also correlated with noninvasive fibrosis stage indicators including liver stiffness [117].
Based on the above findings, serum levels of SLRPs may be novel noninvasive indicative markers for the presence of fibrotic diseases and therapeutic targets in the disease process. Proteoglycans offer new opportunities to evaluate tissue ECM remodeling using tissue or blood samples from patients.
5. Therapeutic Prospect
Decorin-modified mesenchymal stem cells (MSCs-DCN) promoted the expression of IL-10 and IFN-γ in lung tissue 3 months after radiation, reduced the expression of Col1α1 and Col3α1 to normal level, and down-regulated Tregs to inhibit and delay the progression of fibrosis [118]. Minoru shimizukawa et al. found that only intratracheal injection of exogenous DCN increased DCN mRNA expression in lung tissue and decreased hydroxyproline content. In contrast, intravenous injection only increased the expression of DCN in the liver, but not in the lung, and did not reduce pulmonary fibrosis [119]. Due to the degradation of proteases, targeted delivery and effective protein therapy remain a challenge. Vijayan AN et al. prepared PCL-gelatin biomimetic scaffold to optimize biological activity and provide local delivery of recombinant DCN. The biomaterial was biodegradable and provided sustained release of recombinant DCN. In an in vitro fibrosis model, DCN loaded nanofibers could effectively reduce the expression of ECM related proteins [120]. Vascular homing and cell penetrating peptides as targeting carriers, DCN as a therapeutic domain greatly enhanced its biological activity [121]. Utilizing polydopamine membrane to load DCN proteins on titanium surface, the decorating protein coating on titanium surface could inhibit the proliferation and function of fibroblasts and improve the proliferation and function of osteoblasts [122]. After DCN cDNA was transferred into rat skeletal muscle, the expression of DCN in skeletal muscle and kidney increased, which had a significant therapeutic effect on glomerulonephritis induced fibrosis [43]. DCN promoted the binding of low-density lipoprotein particles to collagen, and the injection of bile duct-targeting lecithins-(PC-) coupled DCN (PC-DCN) nanoliposomes via the bile duct alleviated the degree of liver fibrosis and downregulated the liver function indicators [123]. In other respects, DCN also showed preventive effects on epidural fibrosis and epidural adhesions after laminectomy [124]. Pietraszek K et al. identified a 17 amino acid sequence in the LUM core protein, named Lumcorin, that was able to inhibit cell chemotaxis and to replicate the anti-migratory effects of LUM in vitro, exerting anti-tumor effects on melanoma cells, but this peptide has not yet been reported and applied in fibrotic diseases [125].
Based on the existing research results, the combined application of DCN and biomaterials can increase its targeting and biological activity, which is of great significance for the treatment of multi-organ fibrosis. But at the same time, the research on targeted therapy of other members of SLRP family is still poorly understood, partly because SLRP protein has both pro-fibrotic and anti-fibrotic properties, so this part of research still needs to continue to explore.
6. Conclusions
In the previous study, Rockey DC et al. proposed the concept of fibrosis and related medical basis in the pathological remodeling of human multi-organ tissues, and divided the fibrotic response into four stages. The first is that different types of stimuli cause repeated damage to the epithelium, accompanied by a variety of inflammatory responses that stimulate and recruit other cells. The recruitment of inflammatory factors in phase II drives the activation of fibrotic effector cells, while effector cells activated in phase III secrete a large amount of ECM, which affects the process of cell adhesion, proliferation and differentiation. Ultimately, the damage of these cells triggers a variety of pathological effects, promotes excessive tissue repair, and produces a fibrotic response [126]. The role of the various members of the SLRP family in the pan-fibrotic process are complex and puzzling (Figure 5). On the one hand, the increase of SLRP family members is a compensatory mechanism that limits the progression of organ fibrosis. On the other hand, in some fibrotic processes, SLRPs are able to exacerbate fibrosis through pro-fibrotic signaling pathways or by directly affecting collagen synthesis. Considering the importance of fibrotic diseases and the fact that death in many diseases is associated with fibrosis, it is important to further investigate the role of SLRPs.
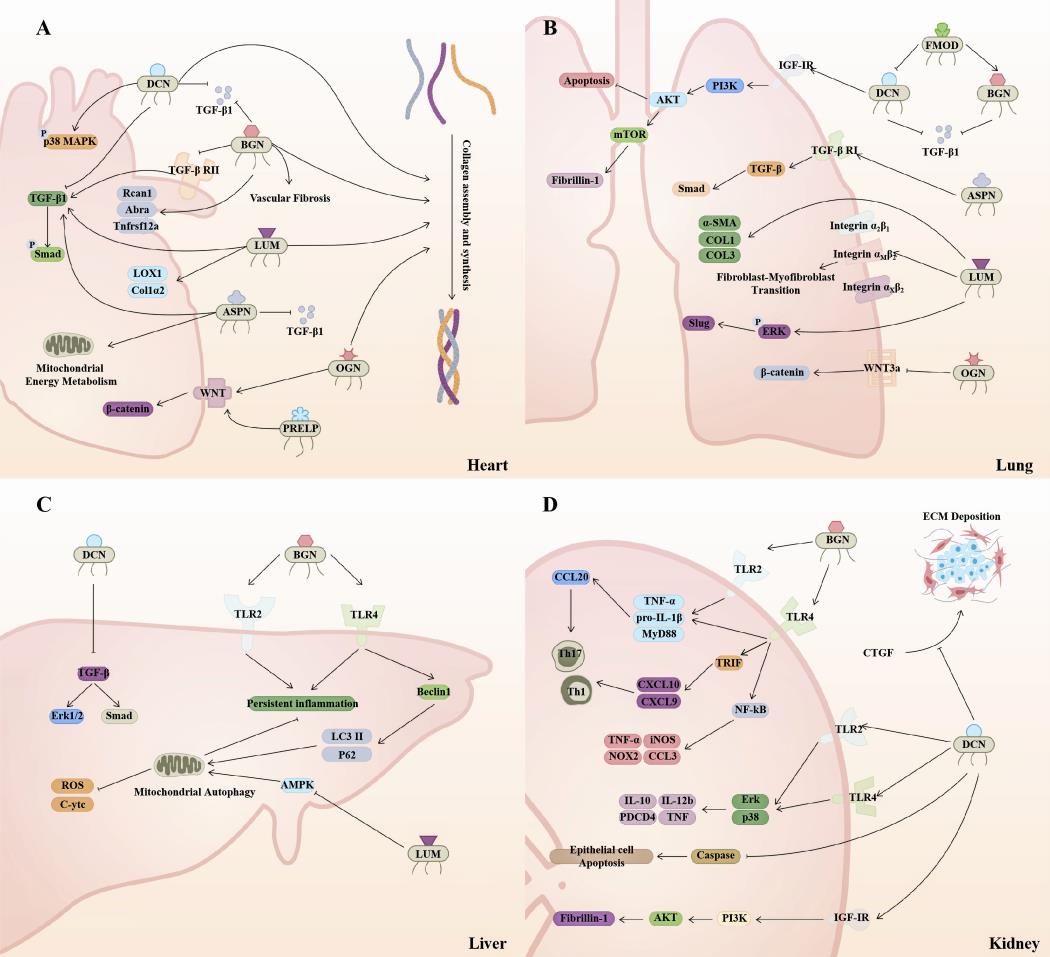
Figure 5. SLRPs are widely involved in the mechanism of multi-organ fibrosis. (A) In the heart, the majority of SLRPs promote collagen assembly and synthesis. Extracellularly, DCN, BGN, and ASPN bind to and inhibit TGF-β1. BGN inhibits TGF-β RII, which together with DCN antagonizes the TGF-β/Smad signaling pathway. In contrast, LUM and ASPN contribute to this pathway. Besides that, OGN and PRELP benefit the Wnt/β-catenin signaling pathway, and DCN promotes p38 MAPK phosphorylation. ASPN promotes mitochondrial energy metabolism. (B) DCN inhibits the TGF-β signaling pathway during liver fibrogenesis. BGN promotes inflammatory responses through TLR2 and TLR4, while also mediating mitophagy to suppress inflammation. LUM decreases AMPK activity and inhibits autophagy while inducing mitochondrial function disruption. (C) DCN and BGN inhibit TGF-β activity. DCN inhibits epithelial cell apoptosis by promoting PI3K/AKT signaling via IGF-IR. ASPN promotes the TGF-β/Smad pathway. LUM mediates fibroblast transdifferentiation via integrin receptors and potentiates the ERK/SLUG pathway. OGN inhibits Wnt/β-catenin signaling to promote fibroblast apoptosis. Meanwhile, FMOD decreases DCN while increasing BGN expression. (D) DCN and BGN promote the expression of downstream inflammatory factors mainly through TLR2 and TLR4, and DCN inhibits tubular epithelial cell apoptosis while mediating PI3K/Akt signaling. In extracellular DCN is able to inhibit CTGF-induced ECM deposition. DCN, decorin; BGN, biglycan; LUM, lumican; FMOD, fibromodulin; OGN, osteoglycin; ASPN, asporin; PRELP, proline and arginine rich end leucine rich repeat protein.
Several recent studies have shown, among others, that SLRPs play a greater role during tumorigenesis, particularly tumor epithelial-mesenchymal transition [127,128]. For example, FMOD secreted by differentiated glioma cells (DGCs) promoted angiogenesis by activating integrin dependent Notch signals in endothelial cells [129]. In addition, FMOD and SOX2 signal coupling positively regulated the brain metastasis and growth of melanoma [130]. BGN participated in the positive feedback loop of FAP and STAT3 to mediate the interaction between tumor cells and mesothelial cells to regulate peritoneal metastasis of gastric cancer [131]. Our current understanding may just touch the surface of several roles of SLRPs in fibrosis and tumorigenesis, with new signaling pathways being discovered each year, and the most important finding of this review is that SLRPs have the potential to be developed as effective diagnostic markers and therapeutic techniques. Based on the above studies, the role and mechanism of SLRPs in multi-organ fibrosis remain to be further explored, which is essential for providing biomarkers and intervention targets.
Author Contributions: Jiayu Guo and Yan Wang wrote original draft. Haihai Liang and Baofeng Yang supervised and drafted manuscript. All authors have read and agreed to the published version of the manuscript.
Funding: This work was supported by the Distinguished Youth Foundation of Heilongjiang Province (JQ2022H001); National Natural Science Foundation of China (32171127); CAMS Innovation Fund for Medical Sciences (CIFMS), 2020-I2M-5-003.
Data Availability Statement: Not applicable.
Conflicts of Interest: The authors declare no conflict of interest.
References
- Hu H. H.; Cao G.; Wu X. Q.; et al. Wnt signaling pathway in aging-related tissue fibrosis and therapies. Ageing Res. Rev., 2020, 60: 101063. doi:10.1016/j.arr.2020.101063. DOI: https://doi.org/10.1016/j.arr.2020.101063
- Kong X.; Liu H.; He X.; et al. Unraveling the Mystery of Cold Stress-Induced Myocardial Injury. Front. Physiol., 2020, 11: 580811. doi:10.3389/fphys.2020.580811. DOI: https://doi.org/10.3389/fphys.2020.580811
- Maldonado H.; Hagood J.S. Cooperative signaling between integrins and growth factor receptors in fibrosis. J. Mol. Med., 2021, 99(2): 213-224. doi:10.1007/s00109-020-02026-2. DOI: https://doi.org/10.1007/s00109-020-02026-2
- Li L.; Zhao Q.; Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol., 2018, 68-69: 490-506. doi:10.1016/j.matbio.2018.01.013. DOI: https://doi.org/10.1016/j.matbio.2018.01.013
- Chaudhuri O.; Cooper-White J.; Janmey P.A.; et al. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature, 2020, 584(7822): 535-546. doi:10.1038/s41586-020-2612-2. DOI: https://doi.org/10.1038/s41586-020-2612-2
- Bonnans C.; Chou J.; Werb Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol., 2014, 15(12): 786-801. doi:10.1038/nrm3904. DOI: https://doi.org/10.1038/nrm3904
- Theocharis A. D.; Skandalis S. S.; Gialeli C.; et al. Extracellular matrix structure. Adv. Drug Deliv. Rev., 2016, 97: 4-27. doi:10.1016/j.addr.2015.11.001. DOI: https://doi.org/10.1016/j.addr.2015.11.001
- Liu C.; Pei M.; Li Q.; et al. Decellularized extracellular matrix mediates tissue construction and regeneration. Front. Med., 2022, 16(1): 56-82. doi:10.1007/s11684-021-0900-3. DOI: https://doi.org/10.1007/s11684-021-0900-3
- Iozzo R. V.; Schaefer L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol., 2015, 42: 11-55. doi:10.1016/j.matbio.2015.02.003 DOI: https://doi.org/10.1016/j.matbio.2015.02.003
- Christensen G.; Herum K. M.; LundeI. G. Sweet, yet underappreciated: Proteoglycans and extracellular matrix remodeling in heart disease. Matrix Biol., 2019, 75-76: 286-299. doi:10.1016/j.matbio.2018.01.001 DOI: https://doi.org/10.1016/j.matbio.2018.01.001
- Westermann D.; Mersmann J.; Melchior A.; et al. Biglycan is required for adaptive remodeling after myocardial infarction. Circulation, 2008, 117(10): 1269-76. doi:10.1161/CIRCULATIONAHA.107.714147 DOI: https://doi.org/10.1161/CIRCULATIONAHA.107.714147
- Prydz K.; Dalen K. T. Synthesis and sorting of proteoglycans. J. Cell Sci., 2000, 113 Pt 2: 193-205. doi:10.1242/jcs.113.2.193. DOI: https://doi.org/10.1242/jcs.113.2.193
- Chen S.; Birk D. E. The regulatory roles of small leucine-rich proteoglycans in extracellular matrix assembly. FEBS J., 2013, 280(10): 2120-37. doi:10.1111/febs.12136. DOI: https://doi.org/10.1111/febs.12136
- Nastase M. V.; Janicova A.; Roedig H.; et al. Small Leucine-Rich Proteoglycans in Renal Inflammation: Two Sides of the Coin. J. Histochem. Cytochem., 2018, 66(4): 261-272. doi:10.1369/0022155417738752. DOI: https://doi.org/10.1369/0022155417738752
- Westergren-Thorsson G.; Hernnäs J.; SärnstrandB.; et al. Altered expression of small proteoglycans, collagen, and transforming growth factor-beta 1 in developing bleomycin-induced pulmonary fibrosis in rats. J. Clin. Invest., 1993, 92(2): 632-7. doi:10.1172/JCI116631. DOI: https://doi.org/10.1172/JCI116631
- Barallobre-Barreiro J.; Gupta S. K.; ZoccaratoA.; et al. Glycoproteomics Reveals Decorin Peptides With Anti-Myostatin Activity in Human Atrial Fibrillation. Circulation, 2016, 134(11): 817-32. doi:10.1161/CIRCULATIONAHA.115.016423 DOI: https://doi.org/10.1161/CIRCULATIONAHA.115.016423
- Meyer D. H.; Krull N.; Dreher K. L. Gressner A. M. Biglycan and decorin gene expression in normal and fibrotic rat liver: cellular localization and regulatory factors. Hepatology, 1992, 16(1): 204-16. doi: 10.1002/hep.1840160131. doi:10.1002/hep.1840160131. DOI: https://doi.org/10.1002/hep.1840160131
- Schaefer L; Mihalik D.; Babelova A.; et al. Regulation of fibrillin-1 by biglycan and decorin is important for tissue preservation in the kidney during pressure-induced injury. Am. J. Pathol., 2004,165(2): 383-96. doi: 10.1016/S0002-9440(10)63305-6. DOI: https://doi.org/10.1016/S0002-9440(10)63305-6
- Stokes M. B.; Holler S.; Cui Y.; et al. Expression of decorin, biglycan, and collagen type I in human renal fibrosing disease. Kidney Int., 2000, 57(2): 487-98. doi: 10.1046/j.1523-1755.2000.00868.x. DOI: https://doi.org/10.1046/j.1523-1755.2000.00868.x
- Shimizu-HirotaR.; SasamuraH.; KurodaM.; et al. Extracellular matrix glycoprotein biglycan enhances vascular smooth muscle cell proliferation and migration. Circ. Res., 2004, 94(8): 1067-74. doi: 10.1161/01.RES.0000126049.79800.CA. DOI: https://doi.org/10.1161/01.RES.0000126049.79800.CA
- Beetz N.; Rommel C.; Schnick T.; et al. Ablation of biglycan attenuates cardiac hypertrophy and fibrosis after left ventricular pressure overload. J. Mol. Cell. Cardiol., 2016, 101: 145-155. doi: 10.1016/j.yjmcc.2016.10.011. Epub 2016 Oct 24. DOI: https://doi.org/10.1016/j.yjmcc.2016.10.011
- Yu M.; He X.; Song X.; et al. Biglycan promotes hepatic fibrosis through activating heat shock protein 47. Liver Int., 2023, 43(2): 500-512. doi: 10.1111/liv.15477. Epub 2022 Nov 21. DOI: https://doi.org/10.1111/liv.15477
- -Maciejewski C. C.; Honardoust D.; Tredget E. E.; et al. Differential expression of class I small leucine-rich proteoglycans in an animal model of partial bladder outlet obstruction. J. Urol., 2012, 188(4 Suppl): 1543-8. doi: 10.1016/j.juro.2012.03.045. Epub 2012 Aug 19. DOI: https://doi.org/10.1016/j.juro.2012.03.045
- Huang C.; Sharma A.; Thakur R.; et al. Asporin, an extracellular matrix protein, is a beneficial regulator of cardiac remodeling. Matrix Biol., 2022, 110: 40-59. doi: 10.1016/j.matbio.2022.04.005. Epub 2022 Apr 22. DOI: https://doi.org/10.1016/j.matbio.2022.04.005
- HuangS.; LaiX.; YangL.; et al. Asporin Promotes TGF-β-induced Lung Myofibroblast Differentiation by Facilitating Rab11-Dependent Recycling of TβRI. Am. J. Respir. Cell Mol. Biol., 2022, 66(2): 158-170. doi: 10.1165/rcmb.2021-0257OC. DOI: https://doi.org/10.1165/rcmb.2021-0257OC
- Qian W.; Xia S.; Yang X.; et al. Complex Involvement of the Extracellular Matrix, Immune Effect, and Lipid Metabolism in the Development of Idiopathic Pulmonary Fibrosis. Front. Mol. Biosci., 2022, 8: 800747. doi: 10.3389/fmolb.2021.800747. DOI: https://doi.org/10.3389/fmolb.2021.800747
- Engebretsen K. V.; Lunde I. G.; Strand M. E.; et al. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. FEBS J., 2013, 280(10): 2382-98. doi: 10.1111/febs.12235. Epub 2013 Apr 2. DOI: https://doi.org/10.1111/febs.12235
- Rixon C.; Andreassen K.; Shen X.; et al. Lumican accumulates with fibrillar collagen in fibrosis in hypertrophic cardiomyopathy. ESC Heart Fail., 2023, 10(2): 858-871. doi: 10.1002/ehf2.14234. Epub 2022 Nov 29. DOI: https://doi.org/10.1002/ehf2.14234
- Mohammadzadeh N.; Melleby A. O.; PalmeroS.; et al. Moderate Loss of the Extracellular Matrix Proteoglycan Lumican Attenuates Cardiac Fibrosis in Mice Subjected to Pressure Overload. Cardiology, 2020, 145(3): 187-198. doi: 10.1159/000505318. Epub 2020 Jan 22. PMID: 31968347; DOI: https://doi.org/10.1159/000505318
- Pilling D.; Vakil V.; Cox N.; et al. TNF-α-stimulated fibroblasts secrete lumican to promote fibrocyte differentiation. Proc. Natl. Acad. Sci. U S A., 2015, 112(38): 11929-34. doi: 10.1073/pnas.1507387112. Epub 2015 Sep 8. DOI: https://doi.org/10.1073/pnas.1507387112
- KrishnanA.; LiX.; KaoW. Y.; et al. Lumican, an extracellular matrix proteoglycan, is a novel requisite for hepatic fibrosis. Lab. Invest., 2012, 92(12): 1712-25. doi: 10.1038/labinvest.2012.121. Epub 2012 Sep 24. DOI: https://doi.org/10.1038/labinvest.2012.121
- Hsieh L. T.; Nastase M. V.; Zeng-Brouwers J.; et al. Soluble biglycan as a biomarker of inflammatory renal diseases. Int. J. Biochem. Cell Biol., 2014, 54: 223-35. doi: 10.1016/j.biocel.2014.07.020. Epub 2014 Aug 1. DOI: https://doi.org/10.1016/j.biocel.2014.07.020
- Xiao D.; Liang T.; Zhuang Z.; et al. Lumican promotes joint fibrosis through TGF-β signaling. FEBS Open Bio., 2020, 10(11): 2478-2488. doi: 10.1002/2211-5463.12974. Epub 2020 Oct 25. DOI: https://doi.org/10.1002/2211-5463.12974
- AndenæsK.; LundeI. G.; MohammadzadehN.; et al. The extracellular matrix proteoglycan fibromodulin is upregulated in clinical and experimental heart failure and affects cardiac remodeling. PLoS One, 2018, 13(7): e0201422. doi: 10.1371/journal.pone.0201422. DOI: https://doi.org/10.1371/journal.pone.0201422
- Soo C.; Hu F. Y.; Zhang X.; et al. Differential expression of fibromodulin, a transforming growth factor-beta modulator, in fetal skin development and scarless repair. Am. J. Pathol., 2000, 157(2): 423-33. doi: 10.1016/s0002-9440(10)64555-5. DOI: https://doi.org/10.1016/S0002-9440(10)64555-5
- Venkatesan N.; Ebihara T.; RoughleyP. J.; et al. Alterations in large and small proteoglycans in bleomycin-induced pulmonary fibrosis in rats. Am. J. Respir. Crit. Care. Med., 2000, 161(6): 2066-73. doi: 10.1164/ajrccm.161.6.9909098. DOI: https://doi.org/10.1164/ajrccm.161.6.9909098
- An W.; Zhu J. W.; Jiang F.; et al. Fibromodulin is upregulated by oxidative stress through the MAPK/AP-1 pathway to promote pancreatic stellate cell activation. Pancreatology., 2020, 20(2): 278-287. doi: 10.1016/j.pan.2019.09.011. Epub 2019 Sep 26. DOI: https://doi.org/10.1016/j.pan.2019.09.011
- WoessnerM. N.; HiamD.; SmithC.; et al. Osteoglycin Across the Adult Lifespan. J. Clin. Endocrinol. Metab., 2022, 107(4): e1426-e1433. doi: 10.1210/clinem/dgab861. DOI: https://doi.org/10.1210/clinem/dgab861
- Van Aelst L. N.; Voss S.; Carai P.; et al. Osteoglycin prevents cardiac dilatation and dysfunction after myocardial infarction through infarct collagen strengthening. Circ. Res., 2015, 116(3): 425-36. doi: 10.1161/CIRCRESAHA.116.304599. Epub 2014 Dec 17. DOI: https://doi.org/10.1161/CIRCRESAHA.116.304599
- Tallquist M. D. Cardiac Fibroblast Diversity. Annu. Rev. Physiol., 2020, 82: 63-78. doi: 10.1146/annurev-physiol-021119-034527. DOI: https://doi.org/10.1146/annurev-physiol-021119-034527
- Frangogiannis N. G. Cardiac fibrosis. Cardiovasc. Res., 2021, 117(6): 1450-1488. doi: 10.1093/cvr/cvaa324. DOI: https://doi.org/10.1093/cvr/cvaa324
- Weis S. M.; Zimmerman S. D.; Shah M.; et al. A role for decorin in the remodeling of myocardial infarction. Matrix Biol., 2005, 24(4): 313-24. doi: 10.1016/j.matbio.2005.05.003. DOI: https://doi.org/10.1016/j.matbio.2005.05.003
- Isaka Y.; Brees D. K.; Ikegaya K.; et al. Gene therapy by skeletal muscle expression of decorin prevents fibrotic disease in rat kidney. Nat. Med., 1996, 2(4): 418-23. doi: 10.1038/nm0496-418. DOI: https://doi.org/10.1038/nm0496-418
- Jahanyar J.; Joyce D. L.; Southard R. E.; et al. Decorin-mediated transforming growth factor-beta inhibition ameliorates adverse cardiac remodeling. J. Heart Lung Transplant., 2007, 26(1): 34-40. doi: 10.1016/j.healun.2006.10.005. Epub 2006 Nov 30. DOI: https://doi.org/10.1016/j.healun.2006.10.005
- Yan W.; Wang P.; Zhao C. X.; et al. Decorin gene delivery inhibits cardiac fibrosis in spontaneously hypertensive rats by modulation of transforming growth factor-beta/Smad and p38 mitogen-activated protein kinase signaling pathways. Hum. Gene Ther., 2009, 20(10): 1190-200. DOI: https://doi.org/10.1089/hum.2008.204
- SchaeferL.; DikicI. Autophagy: Instructions from the extracellular matrix. Matrix Biol., 2021, 100-101: 1-8. doi: 10.1016/j.matbio.2021.06.002. Epub 2021 Jul 2. DOI: https://doi.org/10.1016/j.matbio.2021.06.002
- Goyal A.; Neill T.; Owens R. T.; et al. Decorin activates AMPK, an energy sensor kinase, to induce autophagy in endothelial cells. Matrix Biol., 2014, 34: 46-54. doi: 10.1016/j.matbio.2013.12.011. Epub 2014 Jan 26. DOI: https://doi.org/10.1016/j.matbio.2013.12.011
- Torres A.; Gubbiotti M. A.; Iozzo R. V. Decorin-inducible Peg3 Evokes Beclin 1-mediated Autophagy and Thrombospondin 1-mediated Angiostasis. J. Biol. Chem., 2017, 292(12): 5055-5069. doi: 10.1074/jbc.M116.753632. Epub 2017 Feb 7. DOI: https://doi.org/10.1074/jbc.M116.753632
- Chen C. G.; Gubbiotti M. A.; Kapoor A.; et al. Autophagic degradation of HAS2 in endothelial cells: A novel mechanism to regulate angiogenesis. Matrix Biol., 2020, 90: 1-19. doi: 10.1016/j.matbio.2020.02.001. Epub 2020 Feb 19. DOI: https://doi.org/10.1016/j.matbio.2020.02.001
- Heegaard A. M.; Corsi A.; Danielsen C. C.; et al. Biglycan deficiency causes spontaneous aortic dissection and rupture in mice. Circulation, 2007, 115(21): 2731-8. doi: 10.1161/CIRCULATIONAHA.106.653980. Epub 2007 May 14. DOI: https://doi.org/10.1161/CIRCULATIONAHA.106.653980
- Scuruchi M.; Mannino F.; Imbesi C.; et al. Biglycan Involvement in Heart Fibrosis: Modulation of Adenosine 2A Receptor Improves Damage in Immortalized Cardiac Fibroblasts. Int. J. Mol. Sci., 2023, 24(2): 1784. doi: 10.3390/ijms24021784. DOI: https://doi.org/10.3390/ijms24021784
- Melchior-Becker A.; Dai G.; Ding Z.; et al. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J. Biol. Chem., 2011, 286(19): 17365-75. doi: 10.1074/jbc.M110.192682. Epub 2011 Mar 18. DOI: https://doi.org/10.1074/jbc.M110.192682
- Mohammadzadeh N.; Lunde I. G.; Andenæs K.; et al. The extracellular matrix proteoglycan lumican improves survival and counteracts cardiac dilatation and failure in mice subjected to pressure overload. Sci. Rep., 2019, 9(1): 9206. doi: 10.1038/s41598-019-45651-9. DOI: https://doi.org/10.1038/s41598-019-45651-9
- Chen S. W.; Tung Y. C.; Jung S. M.; et al. Lumican-null mice are susceptible to aging and isoproterenol-induced myocardial fibrosis. Biochem. Biophys. Res. Commun., 2017, 482(4): 1304-1311. doi: 10.1016/j.bbrc.2016.12.033. Epub 2016 Dec 7. DOI: https://doi.org/10.1016/j.bbrc.2016.12.033
- Kalamajski S.; Bihan D.; Bonna A.; et al. Fibromodulin Interacts with Collagen Cross-linking Sites and Activates Lysyl Oxidase. J. Biol. Chem., 2016, 291(15): 7951-60. doi: 10.1074/jbc.M115.693408. Epub 2016 Feb 18. DOI: https://doi.org/10.1074/jbc.M115.693408
- Jazbutyte V.; Fiedler J.; Kneitz S.; et al. MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr), 2013, 35(3): 747-62. doi: 10.1007/s11357-012-9407-9. Epub 2012 Apr 27. DOI: https://doi.org/10.1007/s11357-012-9407-9
- Deckx S.; Heggermont W.; Carai P.; et al. Osteoglycin prevents the development of age-related diastolic dysfunction during pressure overload by reducing cardiac fibrosis and inflammation. Matrix Biol., 2018, 66: 110-124. doi: 10.1016/j.matbio.2017.09.002. Epub 2017 Sep 25. DOI: https://doi.org/10.1016/j.matbio.2017.09.002
- Zuo C.; Li X.; Huang J.; et al. Osteoglycin attenuates cardiac fibrosis by suppressing cardiac myofibroblast proliferation and migration through antagonizing lysophosphatidic acid 3/matrix metalloproteinase 2/epidermal growth factor receptor signalling. Cardiovasc. Res., 2018, 114(5): 703-712. doi: 10.1093/cvr/cvy035. DOI: https://doi.org/10.1093/cvr/cvy035
- Fang Y.; Chang Z.; Xu Z.; et al. Osteoglycin silencing exerts inhibitory effects on myocardial fibrosis and epithelial/endothelial-mesenchymal transformation in a mouse model of myocarditis. Biofactors, 2020, 46(6): 1018-1030. doi: 10.1002/biof.1683. Epub 2020 Nov 3. DOI: https://doi.org/10.1002/biof.1683
- Li X. L.; Yu F.; Li B. Y.; et al. The protective effects of grape seed procyanidin B2 against asporin mediates glycated low-density lipoprotein induced-cardiomyocyte apoptosis and fibrosis. Cell Biol. Int., 2020, 44(1): 268-277. doi: 10.1002/cbin.11229. Epub 2019 Sep 18. DOI: https://doi.org/10.1002/cbin.11229
- Zhang Y.; Fu C.; ZhaoS.; et al. PRELP promotes myocardial fibrosis and ventricular remodelling after acute myocardial infarction by the wnt/β-catenin signalling pathway. Cardiovasc. J. Afr., 2022, 33(5): 228-233. doi: 10.5830/CVJA-2022-001. Epub 2022 Jun 29. DOI: https://doi.org/10.5830/CVJA-2022-001
- Parola M.; Pinzani M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med., 2019, 65: 37-55. doi: 10.1016/j.mam.2018.09.002. Epub 2018 Sep 13. DOI: https://doi.org/10.1016/j.mam.2018.09.002
- Bataller R.; Brenner D. A. Liver fibrosis. J. Clin. Invest. 2005 ;115(2):209-18. doi: 10.1172/JCI24282. Erratum in: J. Clin. Invest., 2005, 115(4):1100. DOI: https://doi.org/10.1172/JCI24282
- Baghy K., Dezso K., Laszlo V., et al. Ablation of the decorin gene enhances experimental hepatic fibrosis and impairs hepatic healing in mice. Lab. Invest. 2010, 439-451.doi:10.1038/labinvest.2010.172 DOI: https://doi.org/10.1038/labinvest.2010.172
- Jang Y. O.; Cho M. Y.; Yun C. O.; et al. Effect of Function-Enhanced Mesenchymal Stem Cells Infected With Decorin-Expressing Adenovirus on Hepatic Fibrosis. Stem Cells Transl. Med., 2016, 5(9): 1247-56. doi: 10.5966/sctm.2015-0323. Epub 2016 Jun 30. DOI: https://doi.org/10.5966/sctm.2015-0323
- Baghy K.; Iozzo R. V.; Kovalszky I. Decorin-TGFβ axis in hepatic fibrosis and cirrhosis. J. Histochem. Cytochem., 2012, 60(4): 262-8. doi: 10.1369/0022155412438104. Epub 2012 Jan 19. DOI: https://doi.org/10.1369/0022155412438104
- LiuJ.; ZhuS.; ZengL.; et al. DCN released from ferroptotic cells ignites AGER-dependent immune responses. Autophagy, 2022, 18(9): 2036-2049. doi: 10.1080/15548627.2021.2008692. Epub 2021 Dec 29. DOI: https://doi.org/10.1080/15548627.2021.2008692
- Schulz M.; Dieh l. V.; Trebicka J.; et al. Biglycan: A regulator of hepatorenal inflammation and autophagy. Matrix Biol., 2021, 100-101: 150-161. doi: 10.1016/j.matbio.2021.06.001. Epub 2021 Jun 10. DOI: https://doi.org/10.1016/j.matbio.2021.06.001
- Roedig H.; Damiescu R.; Zeng-Brouwers J.; et al. Danger matrix molecules orchestrate CD14/CD44 signaling in cancer development. Semin. Cancer Biol., 2020, 62: 31-47. doi: 10.1016/j.semcancer.2019.07.026. Epub 2019 Aug 11. DOI: https://doi.org/10.1016/j.semcancer.2019.07.026
- Zeng-Brouwers J.; Pandey S.; Trebicka J.; et al. Communications via the Small Leucine-rich Proteoglycans: Molecular Specificity in Inflammation and Autoimmune Diseases. J. Histochem. Cytochem., 2020, 68(12): 887-906. doi: 10.1369/0022155420930303. Epub 2020 Jul 6. DOI: https://doi.org/10.1369/0022155420930303
- Li X.; Roife D.; Kang Y.; et al. Extracellular lumican augments cytotoxicity of chemotherapy in pancreatic ductal adenocarcinoma cells via autophagy inhibition. Oncogene, 2016, 35(37): 4881-90. doi: 10.1038/onc.2016.20. Epub 2016 Feb 15. DOI: https://doi.org/10.1038/onc.2016.20
- Ptasinski V. A.; Stegmayr J.; Belvisi M. G.; et al. Targeting Alveolar Repair in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol., 2021, 65(4): 347-365. doi: 10.1165/rcmb.2020-0476TR. DOI: https://doi.org/10.1165/rcmb.2020-0476TR
- UpaguptaC.; ShimboriC.; AlsilmiR.; et al. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev., 2018, 27(148): 180033. doi: 10.1183/16000617.0033-2018. DOI: https://doi.org/10.1183/16000617.0033-2018
- InuiN.; SakaiS.; KitagawaM. Molecular Pathogenesis of Pulmonary Fibrosis, with Focus on Pathways Related to TGF-β and the Ubiquitin-Proteasome Pathway. Int. J. Mol. Sci., 2021, 22(11): 6107. doi: 10.3390/ijms22116107. DOI: https://doi.org/10.3390/ijms22116107
- Kolb M.; Margetts P. J.; Galt T.; et al. Transient transgene expression of decorin in the lung reduces the fibrotic response to bleomycin. Am. J. Respir. Crit. Care. Med., 2001, 163(3 Pt 1): 770-7. doi: 10.1164/ajrccm.163.3.2006084. DOI: https://doi.org/10.1164/ajrccm.163.3.2006084
- KolbM.; MargettsP. J.; SimeP. J.; et al. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol., 2001, 280(6): L1327-34. doi: 10.1152/ajplung.2001.280.6.L1327. DOI: https://doi.org/10.1152/ajplung.2001.280.6.L1327
- Allawadhi P.; Singh V.; Khurana I.; et al. Decorin as a possible strategy for the amelioration of COVID-19. Med. Hypotheses, 2021, 152: 110612. doi: 10.1016/j.mehy.2021.110612. Epub 2021 May 20. DOI: https://doi.org/10.1016/j.mehy.2021.110612
- Nastase M. V.; Iozzo R. V.; Schaefer L. Key roles for the small leucine-rich proteoglycans in renal and pulmonary pathophysiology. Biochim. Biophys. Acta, 2014, 1840(8): 2460-70. doi: 10.1016/j.bbagen.2014.01.035. Epub 2014 Feb 5. DOI: https://doi.org/10.1016/j.bbagen.2014.01.035
- Asakura S.; Kato H.; Fujino S.; et al. Role of transforming growth factor-beta1 and decorin in development of central fibrosis in pulmonary adenocarcinoma. Hum. Pathol., 1999, 30(2): 195-8. doi: 10.1016/s0046-8177(99)90275-7. DOI: https://doi.org/10.1016/S0046-8177(99)90275-7
- Wang K.; Wang Y.; Cao Y.; et al. Lumican is elevated in the lung in human and experimental acute respiratory distress syndrome and promotes early fibrotic responses to lung injury. J. Transl. Med., 2022, 20(1): 392. doi: 10.1186/s12967-022-03597-z. DOI: https://doi.org/10.1186/s12967-022-03597-z
- Shi S.; Li H. Overexpressed microRNA-140 inhibits pulmonary fibrosis in interstitial lung disease via the Wnt signaling pathway by downregulating osteoglycin. Am. J. Physiol. Cell Physiol., 2020, 319(5): C895-C905. doi: 10.1152/ajpcell.00479.2019. Epub 2020 Aug 5. DOI: https://doi.org/10.1152/ajpcell.00479.2019
- Rydell-Törmänen K.; Andréasson K.; Hesselstrand R.; et al. Absence of fibromodulin affects matrix composition, collagen deposition and cell turnover in healthy and fibrotic lung parenchyma. Sci. Rep., 2014, 4: 6383. doi: 10.1038/srep06383. DOI: https://doi.org/10.1038/srep06383
- Li L.; Fu H.; Liu Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. Nat. Rev. Nephrol., 2022, 18(9): 545-557. doi: 10.1038/s41581-022-00590-z. Epub 2022 Jul 4. DOI: https://doi.org/10.1038/s41581-022-00590-z
- LiH.; DixonE. E.; WuH.; et al. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab., 2022, 34(12): 1977-1998.e9. doi: 10.1016/j.cmet.2022.09.026. Epub 2022 Oct 19. DOI: https://doi.org/10.1016/j.cmet.2022.09.026
- Leaf I. A.; Duffield J. S. What can target kidney fibrosis? Nephrol. Dial. Transplant., 2017, 32(suppl_1): i89-i97. doi: 10.1093/ndt/gfw388. DOI: https://doi.org/10.1093/ndt/gfw388
- Stokes M. B.; Hudkins K. L.; Zaharia V.; et al. Up-regulation of extracellular matrix proteoglycans and collagen type I in human crescentic glomerulonephritis. Kidney Int., 2001, 59(2): 532-42. doi: 10.1046/j.1523-1755.2001.059002532.x. DOI: https://doi.org/10.1046/j.1523-1755.2001.059002532.x
- Nastase M. V.; Zeng-Brouwers J.; Beckmann J.; et al. Biglycan, a novel trigger of Th1 and Th17 cell recruitment into the kidney. Matrix Biol., 2018, 68-69: 293-317. doi: 10.1016/j.matbio.2017.12.002. Epub 2017 Dec 15. DOI: https://doi.org/10.1016/j.matbio.2017.12.002
- Nakamura T.; Bonnard B.; Palacios-Ramirez R.; et al. Biglycan Is a Novel Mineralocorticoid Receptor Target Involved in Aldosterone/Salt-Induced Glomerular Injury. Int. J. Mol. Sci., 2022, 23(12): 6680. doi: 10.3390/ijms23126680. DOI: https://doi.org/10.3390/ijms23126680
- Merline R.; Moreth K.; Beckmann J.; et al. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci. Signal., 2011, 4(199): ra75. doi: 10.1126/scisignal.2001868. DOI: https://doi.org/10.1126/scisignal.2001868
- Schaefer L.; Tsalastra W.; Babelova A.; et al. Decorin-mediated regulation of fibrillin-1 in the kidney involves the insulin-like growth factor-I receptor and Mammalian target of rapamycin. Am. J. Pathol., 2007, 170(1): 301-15. doi: 10.2353/ajpath.2007.060497. DOI: https://doi.org/10.2353/ajpath.2007.060497
- Schaefer L.; Macakova K.; Raslik I.; et al. Absence of decorin adversely influences tubulointerstitial fibrosis of the obstructed kidney by enhanced apoptosis and increased inflammatory reaction. Am. J. Pathol., 2002, 160(3): 1181-91. doi: 10.1016/S0002-9440(10)64937-1. DOI: https://doi.org/10.1016/S0002-9440(10)64937-1
- Vial C.; Gutiérrez J.; Santander C.; et al. Decorin interacts with connective tissue growth factor (CTGF)/CCN2 by LRR12 inhibiting its biological activity. J. Biol. Chem., 2011, 286(27):24242-52. doi: 10.1074/jbc.M110.189365. Epub 2011 Mar 23. DOI: https://doi.org/10.1074/jbc.M110.189365
- Zhu J.; Li Y.; Shen W.; et al. Relationships between transforming growth factor-beta1, myostatin, and decorin: implications for skeletal muscle fibrosis. J. Biol. Chem., 2007, 282(35):25852-63. doi: 10.1074/jbc.M704146200. Epub 2007 Jun 27. DOI: https://doi.org/10.1074/jbc.M704146200
- Mohan R. R.; Gupta R.; Mehan M. K.; et al. Decorin transfection suppresses profibrogenic genes and myofibroblast formation in human corneal fibroblasts. Exp. Eye. Res., 2010, 91(2): 238-45. doi: 10.1016/j.exer.2010.05.013. Epub 2010 May 28. DOI: https://doi.org/10.1016/j.exer.2010.05.013
- Liu L.; Yu H.; Long Y.; et al. Asporin inhibits collagen matrix-mediated intercellular mechanocommunications between fibroblasts during keloid progression. FASEB J., 2021, 35(7): e21705. doi: 10.1096/fj.202100111R. DOI: https://doi.org/10.1096/fj.202100111R
- Honardoust D.; Varkey M.; Hori K.; et al. Small leucine-rich proteoglycans, decorin and fibromodulin, are reduced in postburn hypertrophic scar. Wound Repair Regen., 2011, 19(3): 368-78. doi: 10.1111/j.1524-475X.2011.00677.x. Epub 2011 Apr 21. DOI: https://doi.org/10.1111/j.1524-475X.2011.00677.x
- Jiang W.; Ting K.; Lee S.; et al. Fibromodulin reduces scar size and increases scar tensile strength in normal and excessive-mechanical-loading porcine cutaneous wounds. J. Cell. Mol. Med., 2018, 22(4): 2510-2513. doi: 10.1111/jcmm.13516. Epub 2018 Feb 1. DOI: https://doi.org/10.1111/jcmm.13516
- Honardoust D.; Varkey M.; Marcoux Y.; et al. Reduced decorin, fibromodulin, and transforming growth factor-β3 in deep dermis leads to hypertrophic scarring. J. Burn. Care. Res., 2012, 33(2): 218-27. doi: 10.1097/BCR.0b013e3182335980. DOI: https://doi.org/10.1097/BCR.0b013e3182335980
- Andréasson K.; Gustafsson R.; Rydell-Törmänen K.; et al. Limited impact of fibromodulin deficiency on the development of experimental skin fibrosis. Exp. Dermatol., 2016, 25(7): 558-61. doi: 10.1111/exd.13012. DOI: https://doi.org/10.1111/exd.13012
- Zheng Z.; Nguyen C.; Zhang X.; et al. Delayed wound closure in fibromodulin-deficient mice is associated with increased TGF-β3 signaling. J. Invest. Dermatol., 2011, 131(3): 769-78. doi: 10.1038/jid.2010.381. Epub 2010 Dec 30. DOI: https://doi.org/10.1038/jid.2010.381
- Stoff A.; Rivera A. A.; Mathis J. M.; et al. Effect of adenoviral mediated overexpression of fibromodulin on human dermal fibroblasts and scar formation in full-thickness incisional wounds. J. Mol. Med., 2007, 85(5): 481-96. doi: 10.1007/s00109-006-0148-z. Epub 2007 Jan 12. DOI: https://doi.org/10.1007/s00109-006-0148-z
- Liu X. J.; Kong F. Z.; Wang Y. H.; et al. Lumican Accelerates Wound Healing by Enhancing α2β1 Integrin-Mediated Fibroblast Contractility. PLoS One, 2013, 8(6): e67124. doi: 10.1371/journal.pone.0067124. DOI: https://doi.org/10.1371/journal.pone.0067124
- Chacón-Solano E.; León C.; Carretero M.; et al. Mechanistic interrogation of mutation-independent disease modulators of RDEB identifies the small leucine-rich proteoglycan PRELP as a TGF-β antagonist and inhibitor of fibrosis. Matrix Biol., 2022, 111: 189-206. doi: 10.1016/j.matbio.2022.06.007. Epub 2022 Jun 30. DOI: https://doi.org/10.1016/j.matbio.2022.06.007
- Moreth K.; Brodbeck R.; Babelova A.; et al. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J. Clin. Invest., 2010, 120(12): 4251-72. doi: 10.1172/JCI42213. Epub 2010 Nov 15. DOI: https://doi.org/10.1172/JCI42213
- Nikaido T.; Tanino Y.; Wang X.; et al. Serum decorin is a potential prognostic biomarker in patients with acute exacerbation of idiopathic pulmonary fibrosis. J. Thorac. Dis., 2018, 10(9): 5346-5358. doi: 10.21037/jtd.2018.08.60. DOI: https://doi.org/10.21037/jtd.2018.08.60
- Kehlet S. N.; Bager C. L.; Willumsen N.; et al. Cathepsin-S degraded decorin are elevated in fibrotic lung disorders - development and biological validation of a new serum biomarker. BMC Pulm. Med., 2017, 17(1): 110. doi: 10.1186/s12890-017-0455-x. DOI: https://doi.org/10.1186/s12890-017-0455-x
- Ueland T.; Aukrust P.; Nymo S. H.; et al. Novel extracellular matrix biomarkers as predictors of adverse outcome in chronic heart failure: association between biglycan and response to statin therapy in the CORONA trial. J. Card. Fail., 2015, 21(2): 153-9. doi: 10.1016/j.cardfail.2014.10.016. Epub 2014 Nov 7. DOI: https://doi.org/10.1016/j.cardfail.2014.10.016
- Cengiz M.; Yilmaz G.; Ozenirler S. Serum Biglycan as a Diagnostic Marker for Non-Alcoholic Steatohepatitis and Liver Fibrosis. Clin. Lab., 2021, 67(3). doi: 10.7754/Clin.Lab.2020.200709. DOI: https://doi.org/10.7754/Clin.Lab.2020.200709
- Ciftciler R.; Ozenirler S.; Yucel A. A.; et al. The importance of serum biglycan levels as a fibrosis marker in patients with chronic hepatitis Br. J. Clin. Lab. Anal., 2017, 31(5): e22109. doi: 10.1002/jcla.22109. Epub 2016 Dec 7. DOI: https://doi.org/10.1002/jcla.22109
- Romero R.; Mazaki-Tovi S.; Vaisbuch E.; et al. Metabolomics in premature labor: a novel approach to identify patients at risk for preterm delivery. J. Matern. Fetal Neonatal Med., 2010, 23(12): 1344-59. doi: 10.3109/14767058.2010.482618. Epub 2010 May 26. DOI: https://doi.org/10.3109/14767058.2010.482618
- Cheng J. M.; Akkerhuis K. M.; Meilhac O.; et al. Circulating osteoglycin and NGAL/MMP9 complex concentrations predict 1-year major adverse cardiovascular events after coronary angiography. Arterioscler, Thromb, Vasc, Biol., 2014, 34(5): 1078-84. doi: 10.1161/ATVBAHA.114.303486. Epub 2014 Mar 20. DOI: https://doi.org/10.1161/ATVBAHA.114.303486
- Motiwala S. R.; Szymonifka J.; Belcher A.; et al. Measurement of novel biomarkers to predict chronic heart failure outcomes and left ventricular remodeling. J. Cardiovasc. Transl. Res., 2017, 250-61. doi: 10.1007/s12265-013-9522-8. Epub 2013 Dec 6. DOI: https://doi.org/10.1007/s12265-013-9522-8
- Deckx S.; Heymans S.; Papageorgiou A. P. The diverse functions of osteoglycin: a deceitful dwarf, or a master regulator of disease? FASEB J., 2016, 30(8):2651-61. doi: 10.1096/fj.201500096R. Epub 2016 Apr 14. DOI: https://doi.org/10.1096/fj.201500096R
- Baek S. H.; Cha R. H.; Kang S. W.; et al. Higher Serum Levels of Osteoglycin Are Associated with All-Cause Mortality and Cardiovascular and Cerebrovascular Events in Patients with Advanced Chronic Kidney Disease. Tohoku. J. Exp. Med., 2017, 242(4): 281-290. doi: 10.1620/tjem.242.281. DOI: https://doi.org/10.1620/tjem.242.281
- Hsu M. E.; Cheng Y. T.; Chang C. H.; et al. Level of serum soluble lumican and risks of perioperative complications in patients receiving aortic surgery. PLoS One, 2021, 16(3): e0247340. doi: 10.1371/journal.pone.0247340. DOI: https://doi.org/10.1371/journal.pone.0247340
- Gu G.; Wan F.; Xue Y.; et al. Lumican as a novel potential clinical indicator for acute aortic dissection: A comparative study, based on multi-slice computed tomography angiography. Exp. Ther. Med., 2016, 11(3): 923-928. doi: 10.3892/etm.2016.3020. Epub 2016 Jan 22. DOI: https://doi.org/10.3892/etm.2016.3020
- Decaris M. L.; Li K. W.; Emson C. L.; et al. Identifying nonalcoholic fatty liver disease patients with active fibrosis by measuring extracellular matrix remodeling rates in tissue and blood. Hepatology, 2017, 65(1): 78-88. doi: 10.1002/hep.28860. Epub 2016 Nov 15. DOI: https://doi.org/10.1002/hep.28860
- Liu D.; Kong F.; Yuan Y.; et al. Decorin-Modified Umbilical Cord Mesenchymal Stem Cells (MSCs) Attenuate Radiation-Induced Lung Injuries via Regulating Inflammation, Fibrotic Factors, and Immune Responses. Int. J. Radiat. Oncol. Biol. Phys., 2018, 101(4) 945-956. doi: 10.1016/j.ijrobp.2018.04.007. Epub 2018 Apr 16.
- Shimizukawa M.; Ebina M.; Narumi K.; et al. Intratracheal gene transfer of decorin reduces subpleural fibroproliferation induced by bleomycin. Am. J. Physiol. Lung Cell. Mol. Physiol., 2003, 284(3): L526-32. doi: 10.1152/ajplung.00131.2002. DOI: https://doi.org/10.1152/ajplung.00131.2002
- Vijayan A. N.; Solaimuthu A.; Murali P.; et al. Decorin mediated biomimetic PCL-gelatin nano-framework to impede scarring. Int. J. Biol. Macromol., 2022, 219: 907-918. doi: 10.1016/j.ijbiomac.2022.08.029. Epub 2022 Aug 8. DOI: https://doi.org/10.1016/j.ijbiomac.2022.08.029
- Järvinen T. A. H.; Ruoslahti E. Generation of a multi-functional, target organ-specific, anti-fibrotic molecule by molecular engineering of the extracellular matrix protein, decorin. Br. J. Pharmacol., 2019, 176(1): 16-25. doi: 10.1111/bph.14374. Epub 2018 Jun 25. DOI: https://doi.org/10.1111/bph.14374
- He R.; Lu Y.; Ren J.; et al. Decreased fibrous encapsulation and enhanced osseointegration in vitro by decorin-modified titanium surface. Colloids Surf. B Biointerfaces, 2017, 155: 17-24. doi: 10.1016/j.colsurfb.2017.03.055. Epub 2017 Mar 31. DOI: https://doi.org/10.1016/j.colsurfb.2017.03.055
- Chen G.; Zhu Y.; Liang X.; et al. The Effect of Lecithins Coupled Decorin Nanoliposomes on Treatment of Carbon Tetrachloride-Induced Liver Fibrosis. Biomed Res. Int., 2020, 8815904. doi: 10.1155/2020/8815904. DOI: https://doi.org/10.1155/2020/8815904
- Ding Q.; Wei Q.; Sheng G.; et al. The Preventive Effect of Decorin on Epidural Fibrosis and Epidural Adhesions After Laminectomy. Front. Pharmacol., 2021, 12: 774316. doi: 10.3389/fphar.2021.774316. DOI: https://doi.org/10.3389/fphar.2021.774316
- Pietraszek K.; Brézillon S.; Perreau C.; et al. Lumican - derived peptides inhibit melanoma cell growth and migration. PLoS One, 2013, 8(10):e76232. doi: 10.1371/journal.pone.0076232. DOI: https://doi.org/10.1371/journal.pone.0076232
- Rockey D. C.; Bell P. D.; Hill J. A. Fibrosis--a common pathway to organ injury and failure. N. Engl. J. Med., 2015, 372(12): 1138-49. doi: 10.1056/NEJMra1300575. DOI: https://doi.org/10.1056/NEJMra1300575
- Mao L.; Yang J.; Yue J.; et al. Decorin deficiency promotes epithelial-mesenchymal transition and colon cancer metastasis. Matrix Biol., 2021, 95: 1-14. doi: 10.1016/j.matbio.2020.10.001. Epub 2020 Oct 13. DOI: https://doi.org/10.1016/j.matbio.2020.10.001
- Karamanou K.; Franchi M.; Vynios D; et al. Epithelial-to-mesenchymal transition and invadopodia markers in breast cancer: Lumican a key regulator. Semin. Cancer Biol., 2020, 62: 125-133. doi: 10.1016/j.semcancer.2019.08.003. Epub 2019 Aug 8. DOI: https://doi.org/10.1016/j.semcancer.2019.08.003
- Sengupta S.; Mondal M.; Prasasvi K. R.; et al. Differentiated glioma cell-derived fibromodulin activates integrin-dependent Notch signaling in endothelial cells to promote tumor angiogenesis and growth. Elife, 2022, 11: e78972. doi: 10.7554/eLife.78972. DOI: https://doi.org/10.7554/eLife.78972
- Oria V. O.; Zhang H.; Zito C. R.; et al. Coupled fibromodulin and SOX2 signaling as a critical regulator of metastatic outgrowth in melanoma. Cell. Mol. Life Sci., 2022, 79(7): 377. doi: 10.1007/s00018-022-04364-5. DOI: https://doi.org/10.1007/s00018-022-04364-5
- Wu H.; Xiang Z.; Huang G.; et al. BGN/FAP/STAT3 positive feedback loop mediated mutual interaction between tumor cells and mesothelial cells contributes to peritoneal metastasis of gastric cancer. Int. J. Biol. Sci., 2023, 19(2): 465-483. doi: 10.7150/ijbs.72218. DOI: https://doi.org/10.7150/ijbs.72218


