

Downloads
Download



This work is licensed under a Creative Commons Attribution 4.0 International License.
Article
Oxidized LDL Regulates Endothelin-1 and Oxidative Stress in Vascular Endothelial Cells: Role of Extracellular Regulated Kinase1/2 (ERK1/2)
Haishan Xu 1,#, Jinhong Duan 1,#, Jun Tao 2, Wen Wang 3, Yunqing Wu 1,^, Shunling Dai 1,*, and Jun Ren 4,5,
1 Faculty of Basic Medicine, Peking Union Medical College, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Beijing 100005 China
2 Department of Cardiovascular Surgery, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou 510000 China
3 Department of Pathophysiology, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China
4 Department of Cardiology, Zhongshan Hospital Fudan University, Shanghai 200032, China
5 National Clinical Research Center for Interventional Medicine, Shanghai 200032, China
# These two authors contributed equally to this work
^ Deceased
* Correspondence: daishunling@aliyun.com (Shunling Dai); jren_aldh2@outlook.com (Jun Ren)
Received: 8 February 2023
Accepted: 7 March 2023
Published: 27 June 2023
Abstract: It is perceived that oxidized low density lipoprotein (oxLDL) perturbs endothelial function and fosters endothelin-1 (ET-1) secretion although the underlying mechanism remains elusive. This study was designed to decipher potential mechanisms underscoring oxLDL-evoked regulation of ET-1 and signaling pathways involved in endothelial cells. ET-1 mRNA expression, secretion and promoter function were determined using RT-PCR, enzyme immunometric and luciferase assays, respectively. GO and GSEA bioinformatics analyses depicted differentially expressed genes (DEGs) mainly associated with cell proliferation, cell division, cellular structure, energy supply, and apoptosis in oxLDL-challenged endothelial cells. Incubation of oxLDL overtly increased ROS production, apoptosis, mRNA level, secretion and promoter activity of ET-1 in human umbilical vein endothelial cells (HUVECs), the effects were mitigated by N-Acetyl Cysteine (NAC). Moreover, oxLDL challenge evoked phosphorylation of extracellular signal-regulated kinase1/2 (ERK1/2) in HUVECs, the effect was reversed by NAC and MEK inhibitor PD98059. NAC and PD98059 nullified oxLDL- induced rises in mRNA expression, secretion and promoter activity of ET-1. Truncation of 5’-flanking sequence of ET-1 (–566 bpLuc to –250 bpLuc) displayed elevated luciferase activity with 24-h oxLDL incubation. Fusion plasmid from –233 and –185 bp Luc drastically dampened luciferase activity in basal and oxLDL-challenged HUVECs. Transfection of reporter construct –250 bp Luc with a 2 bp mutation at AP-1 locus, removed basal and oxLDL- evoked rises in ET-1 promoter activity. Collectively, our findings support that oxLDL evoked activation of ERK1/2 signaling likely through ROS production, en route to upregulation of endothelial transcriptional factor AP-1, resulting in expression and secretion of ET-1.
Keywords:
oxLDL ROS ET-1 stress signaling transcriptional factor endothelial cells1. Introduction
Endothelial injury has been well perceived to evoke pathophysiological anomalies in cardiovascular diseases including coronary heart disease, atherosclerosis, obesity, diabetes, hypertension, and stroke [1–4]. Among numerous etiological cues for endothelial dysfunction, endothelin-1 (ET-1), a polypeptide with 21 amino acids, has received much attention over the past decades as the most potent and long-acting vasoconstrictive peptide [2,5]. ET-1 levels are elevated in various vascular and/or endothelial injuries to foster inflammation, endothelial adhesion, proliferation and migration of vascular smooth muscle cells (VSMCs) to induce onset and development of hypertension, atherosclerosis, pulmonary hypertension and chronic kidney disease [5,6]. Not surprisingly, ET-1 system has become a rather promising target for therapeutic interventions of human diseases with many candidate drugs on the market [4,7]. Clinical trials continue to surface on the utilization of endothelin receptor antagonists in the management of cardiovascular diseases [7]. Among various pathological regulators for ET-1, oxidized low density lipoprotein (oxLDL) is deemed one of the main driving forces for endothelial injury and atherosclerosis, working independently or synergistically with ET-1 [8–10]. Earlier evidence from our lab and others has demonstrated the contribution of oxLDL in promoting gene expression and secretion of ET-1 [8,11–13], although detailed governing pathways for oxLDL remain elusive in vascular endothelial cells.
Reactive oxygen species (ROS), including superoxide (O2-) and hydrogen peroxide (H2O2), serve as essential signaling molecules for the onset and progression of cardiovascular diseases [14–19]. Excess ROS accumulation instigates onset and development of endothelial dysfunction and generation of mitogenic factors, resulting in hyperproliferation of smooth muscle cells and vascular plaque formation [2,5]. Other than its cellular damaging role, ROS may also function as vivid second messengers to govern intracellular signal transduction and transcription factors such as activator protein-1 (AP-1) [20,21]. Of note, ROS and oxidative stress have been demonstrated to evoke elevated ET-1 gene expression in cardiac fibroblasts and vascular endothelial cells [10,22–24]. Nonetheless, whether ROS and oxidative stress participate in oxLDL-induced changes in ET-1 gene expression remains unclear in vascular endothelial cells. To this end, this study was designed to discern the regulatory machinery of oxLDL-induced ET-1 gene expression, if any, in vascular endothelial cells with an emphasis on ROS production, stress signaling and transcription factors. Here oxLDL was employed as the pro-oxidant stressor predominantly based on its pathological impact in the etiology of endothelial dysfunction and atherosclerosis [11,12]. Our results suggested an essential role for ROS in oxLDL-evoked induction of ET-1 gene expression in vascular endothelial cells. Moreover, our data favored a crucial role for redox-sensitive transcription factor AP-1 and extracellular signal-regulated kinase 1/2 (ERK1/2) in oxLDL-evoked ET-1 gene expression.
2. Materials and Methods
2.1. Gene Ontology (GO) Analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis, Gene Set Enrichment Analysis (GSEA) and Visualization
Raw data of GSE137578 from Gene Expression Omnibus (GEO), including ox-LDL-treated human aortic endothelial cells (HAECs) and untreated HAECs (n=3), were imported and analyzed in R studio using the Bioconductor LIMMA (Linear models for microarray and RNA-Seq data) package [25]. Thresholds for differentially expressed genes (DEGs) (ox-LDL treated versus nontreated HAECs) were set as a |log2 fold change > 1.00 with a p value < 0.05. GO enrichment analysis and KEGG pathway analysis were performed to determine significantly enriched terms/pathways of DEGs in ox-LDL-treated versus nontreated HAECs using the R package cluster Profiler and KEGG database [26,27]. Bubble plots displaying the enriched GO terms and KEGG pathways for the DEGs were created using the GOplot R-Package [28]. GESA was performed using online tools available at http://software.broadinstitute.org/gsea [29], which analyzed the entire raw matrix of GSE137578. Visualization was performed at https://www.omicshare.com/tools.
2.2. Human Umbilical Vein Endothelial Cells (HUVECs) and Drug Treatment
HUVEC cells obtained from ATCC (Manassas, VA, USA) were cultured using Medium 199 (10 mM HEPES, 1 mM glutamine, 0.1 mg/mL heparin, 100 µg/mL endothelial cell growth supplement, 100 unit/mL penicillin and 100 µg/mL streptomycin, pH 7.4) supplemented with 10 % fetal bovine serum at 37oC with 5% CO2 with a density of ~105 cells/mL. The second through the fifth passages of confluent HUVEC cells were challenged with oxLDL (35 µg/mL) for 24 h in the absence or presence of antioxidant NAC (10 µM) [30] or the MEK inhibitor PD98059 (30 µM) [30] prior to biochemical assessment. The dose (35 μg/mL) and duration (24 h) chosen for oxLDL were mainly based on our previous finding [12].
2.3. ROS Measurement
Intracellular oxidation dye 2,7-dichlorofluorescin fluorescence (DCF) which converts 2,7-dichlorofluorescin into fluorescent DCF was used as an intracellular oxidation probe. HUVECs challenged with oxLDL, for 24 h in the absence or presence of antioxidant NAC, or the MEK inhibitor PD98059 were loaded with DCF diacetate (25 µM) for 50 min. Fluorescence intensity was monitored using fluorescence microscopic imaging with excitation and emission wavelengths at 488 nm and 510 nm, respectively [30]. DCF procedure was performed in the dark to minimize photobleaching.
2.4. Caspase-3 Assay
Activity of caspase-3, an enzyme activated with apoptosis, was evaluated as a marker for apoptosis. HUVECs were centrifuged at 10,000x g for 10 min, and pellets were lysed in an ice-cold lysis buffer containing HEPES (50 mM), CHAPS (0.1%), dithiothreitol (1 mM), EDTA (0.1 mM), and NP40 (0.1%). Then reaction buffer was supplied followed by addition of caspase-3 colorimetric substrate (Ac-DEVD-pNA) and was incubated at 37°C for 1 h. Caspase-3 activity (microplate reading at 405 nm) was shown as picomoles of pNA released per microgram of protein per minute [31].
2.5. ET-1 Measurement
ET-1 levels were assessed using a human ET-1 enzyme immunometric assay kit (Assay Designs, Inc. Ann Arbor, MI, USA) based on a double-antibody sandwich technique [30,32].
2.6. RNA Isolation and RT-PCR Amplification
Total RNA was isolated from HUVECs using TRIzol (Invitrogen, Carlsbad, CA, USA) and was subjected to DNAase treatment prior to RT-PCR measurement. First strand cDNA templates were created in a reaction condition including 2 µg of total RNA, 500 ng of oligo(dT), 10 mM dNTPs, 100 mM dithiothreitol, and 50 units of superscript reverse transcriptase (Invitrogen). Primers were synthesized using CGTTGTTCCGTATGGACTTG (ET-1, sense), AGGCTATGGCTTCAGACAGG (ET-1, antisense); ACGGATTTGGTCGTATTGGG (GAPDH, sense), TCCTGGAAGATGGTGATGGG (GAPDH, antisense). Following denaturing at 94℃ for 5 min, PCR amplification was conducted for 30 cycles (94℃ for 30 s, 53℃ for 30 s and 72℃ for 60 s), with a final extension step (72℃ for 10 min). The housekeeping gene GAPDH was used as an internal control [30,33].
2.7. Western Blot Analysis
HUVEC cells were lysed in 20 mM Tris (pH 7.4) containing 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X, 0.1% SDS, 1% sodium deoxychlorate, 1 mM phenylmethylsulfonyl fluoride, 50 mM sodium fluoride, 1 mM sodium orthvanadate, 50 µg/mL aprotinin and 50 µg/mL leupeptin. Thirty µg protein sample was loaded to 10% SDS-polyacrylamide gel. Proteins were transferred to PVDF membranes (BioRad, Hercules, CA, USA). Membranes were incubated overnight at 4°C with the anti-ERK1/2, anti-pERK1/2, and anti-AP-1 antibodies. Following rinsing, membranes were incubated with specific secondary antibodies for 2 h at room temperature. Gel band density was detected using Super-Signal West Dura Extended Duration Substrate (Pierce Co. Rockford, IL, USA) [24,33].
2.8. ET-1 Reporter Gene Construction
ET-1 promoter was constructed as follows: pGL3-ET-1-431/+135, pGL3-ET-1-293/+135, pGL3-ET-1-149/+135, pGL3-ET-1-115/+135, pGL3-ET-1-98/+135 and pGL3-ET-1-50/+135. The pGL3-vector was obtained from Promega (Madison, WI, USA). The corresponding sequences in the fragment were 566 bp, 428 bp, 284 bp, 250 bp, 233 bp and 185 bp of the 5’-flanking regions of ET-1 gene, respectively. Reporter plasmids with mutation in the AP-1 site (mutAP-1) were derived from pGL3-ET-1-115/+135 (250 bp) using PCR-based site directed mutagenesis. In the mutAP-1 reporter, the native sequence GTGACTAA was mutated to GGTACTAA [30].
2.9. Transfection and Luciferase Assays
HUVECs were transfected with 2 µg of the appropriate luciferase construct plasmid DNA (native or mutant) using the lipofectin method (Invitrogen). To adjust differences in transfection efficiency, 1 µg of pSV-β-galactosidase plasmid DNA was co-transfected in all experiments. Treatment with various chemicals such as NAC and PD98059 was conducted after 48 h of transfection. The ratio of luciferase-to-β-gal activity was expressed as the relative luciferase activity. The luciferase and β-galactosidase enzyme assay kits were purchased from Promega (Madison, WI, USA) [30].
2.10. Statistical Analysis
Data were shown as mean ± SEM. Statistical analysis was carried out using the one-way analysis of variance (ANOVA) followed by a Tukey post hoc analysis. A value of p < 0.05 was deemed statistically significant.
3. Results
3.1. GO and GSEA Analysis of GEO Database for DEGs for Ox-LDL Challenge
Functional GO analysis revealed that DEGs were significantly enriched among "cell division", "mitotic cell cycle", "DNA replication", "mitotic spindle organizatio" and "chromosome segregation" with respects to biological process, indicating a possible role of ox-LDL in cell proliferation regulation (Figure 1A). For cellular component analysis, DEGs were overtly enriched among "cytosol", "nucleoplasm", "cytoplasm", "nucleus" and "centrosome" (Figure 1B). Besides, DEGs were enriched in "protein binding", "single-stranded DNA-dependent ATP-dependent DNA helicase activity", "microtubule binding", "identical protein binding" and "ATP binding" on molecular function, which are closely related to cellular structural regulation and energy supply (Figure 1C). Moreover, these DEGs possess a direct correlation with "cell cycle", "DNA replication", "pathways in cancer", "p53 signaling pathway" and "rheumatoid arthritis" according to the KEGG pathway database, indicating a connection to cell apoptosis and cell division (Figure 1D).
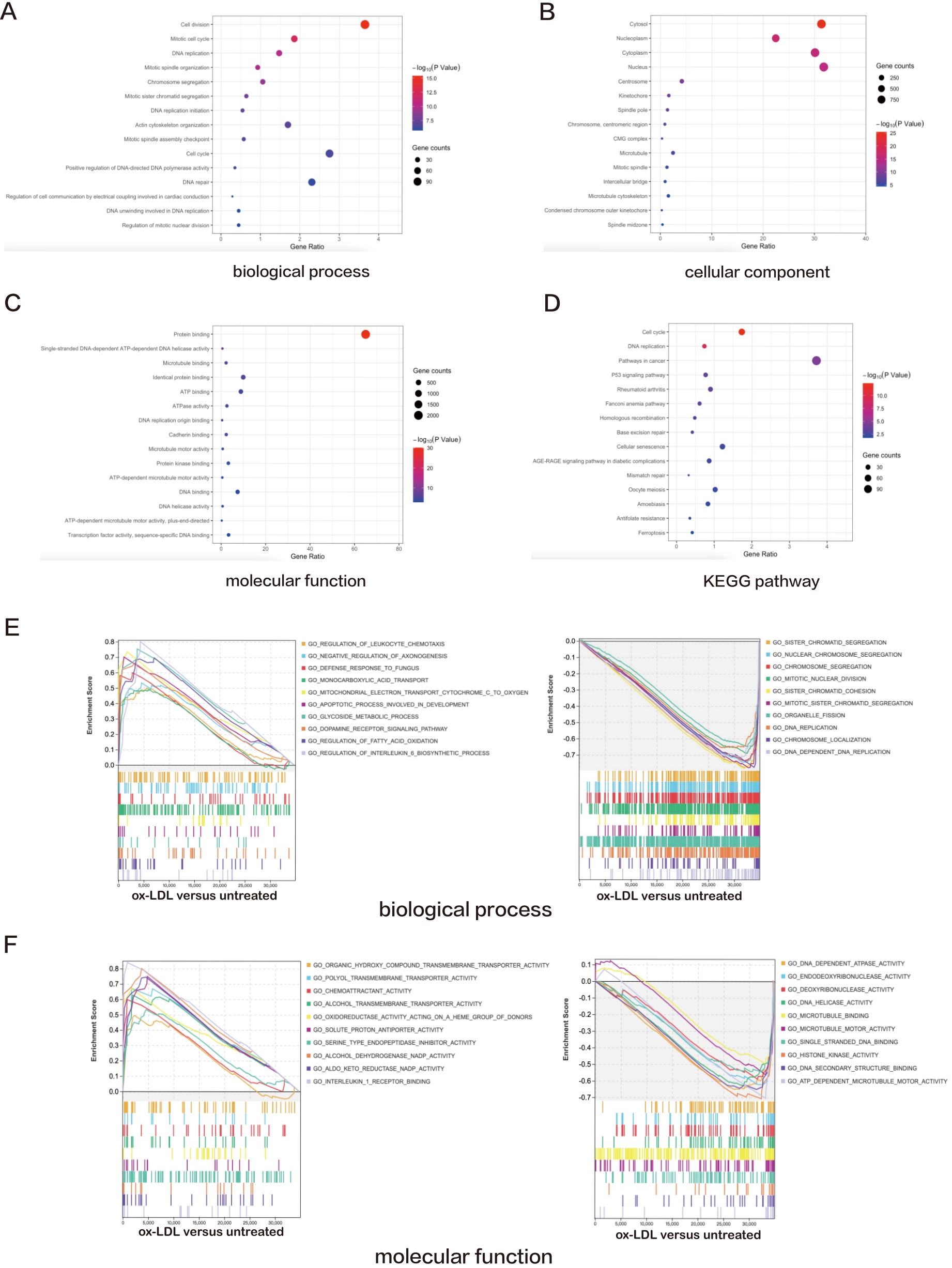
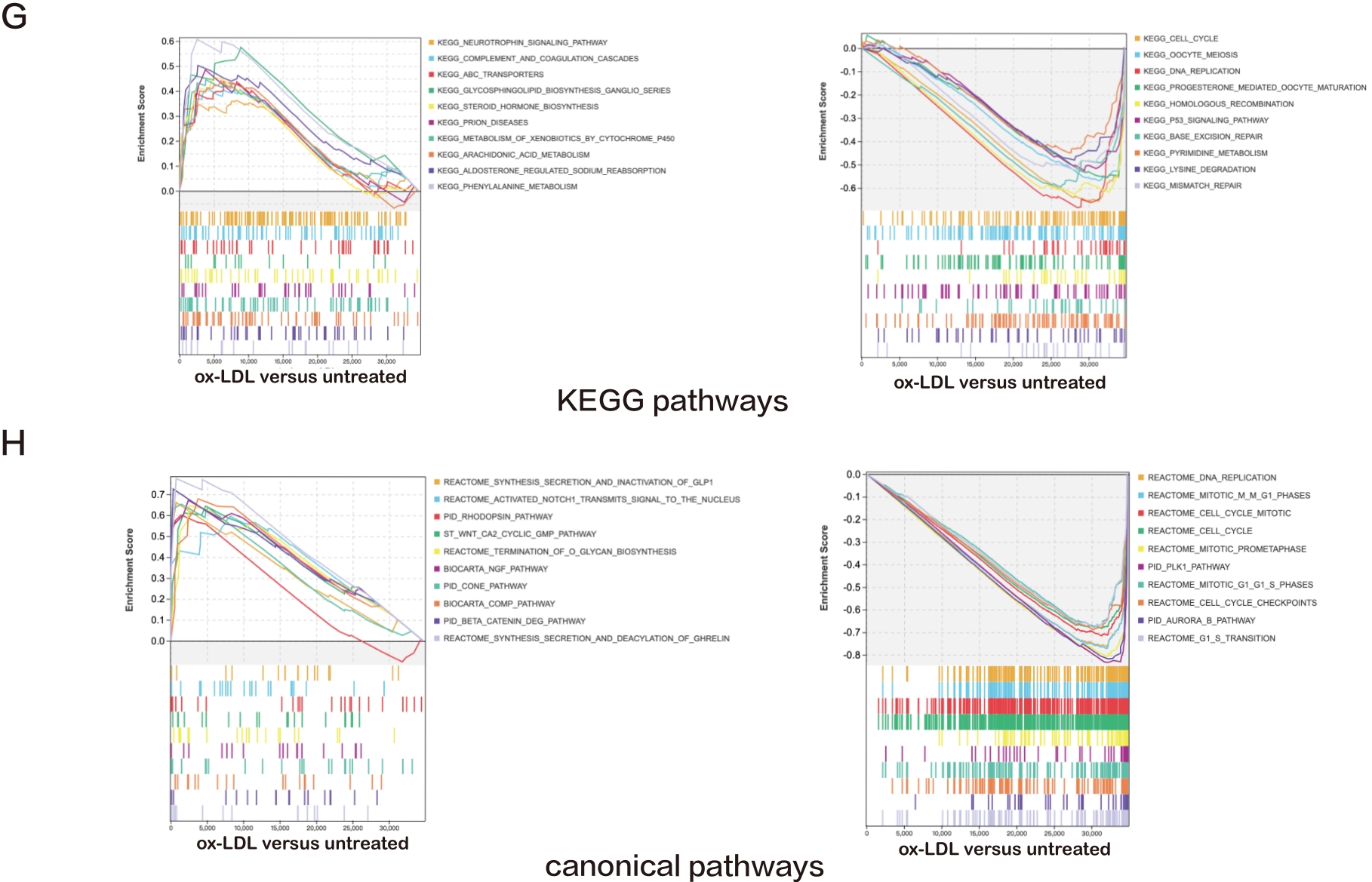
Figure 1. A—D: Bubble plots exhibiting the results of GO and KEGG pathway analyses for the DEGs in ox-LDL treated HAECs versus untreated HAECs. The top 15 significant GO terms and KEGG pathways are displayed; E—H: Line charts displaying results of GSEA for the entire raw matrix data in ox-LDL-treated HAECs versus untreated HAECs. The top 10 significant GO terms, KEGG pathways and canonical pathways are displayed.
Furthermore, GSEA was performed. The most abundantly upregulated biological process terms included "apoptotic process involved in development", "glycoside metabolic process", "dopamine receptor signaling", "regulation of fatty acid oxidation", "regulation of interleukin 6 biosynthetic process", and the most downregulated pathways included "sister chromatid segregation", "nuclear chromosome segregation", "chromosome segregation", "mitotic nuclear division", and "sister chromatid cohesion" (Figure 1E). The most upregulated molecular function terms included "solute proton antiporter activity", "serine type endopeptidase inhibitor activity", "alcohol dehydrogenase NADP activity", "aldo keto reductase NADP activity", and "interleukin 1 receptor binding", and the most downregulated included "DNA dependent ATPase activity", "endo-deoxyribonuclease activity", "deoxyribonuclease activity", "DNA helicase activity", and "microtubule binding" (Figure 1F).
The most upregulated KEGG pathways included "prion diseases", "metabolism of xenobiotics by cytochrome p450", "arachidonic acid metabolism", "aldosterone regulated sodium reabsorption", and "phenylalanine metabolism", while the most downregulated included "cell cycle", "oocyte meiosis", "DNA replication", "progesterone mediated oocyte maturation", and "homologous recombination" (Figure 1G). The most upregulated canonical pathways included "NGF pathway", "cone pathway", "comp pathway", "beta catenin deg pathway", and "synthesis secretion and deacylation of ghrelin", while the most downregulated included "DNA replication", "mitotic m-m G1 phases", "cell cycle mitotic", "cell cycle", and "mitotic prometaphase" (Figure 1H).
3.2. Role of ROS in oxLDL-induced ET-1 Gene Expression and Secretion
oxLDL was shown to provoke ROS production and apoptosis in vascular endothelial cells [34]. Using the fluorescent dye DCF and caspase-3 activity assay, our results noted that oxLDL overtly increased intracellular ROS levels and caspase-3 activity in HUVECs, the effects of which were mitigated by the antioxidant NAC (10 µM) with little effect from NAC itself (p < 0.001, Figure 2A—2C). 24 h incubation of oxLDL drastically enhanced mRNA expression and secretion of ET-1 in HUVECs, the effects of which were abrogated by NAC with minimal effect from NAC itself (p < 0.0001, Figure 2D—E). These findings revealed a vital role for intracellular ROS generation and possibly apoptosis in oxLDL-induced rises in ET-1 levels in vascular endothelial cells.
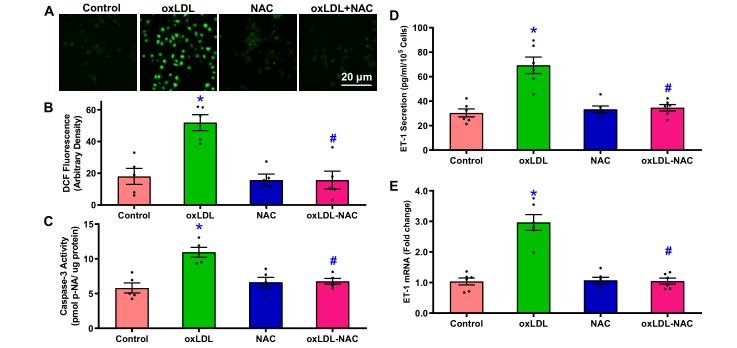
Figure 2. ROS generation, apoptosis, ET-1 secretion and mRNA expression in HUVECs challenged with oxLDL (35 µg/mL) for 24 h in the absence or presence of the antioxidant NAC (10 µM). A: Representative DCF fluorescence images; B: Pooled data of ROS generation; C: Caspase-3 activity; D: ET-1 secretion; and E: ET-1 mRNA expression. Mean ± SEM, n = 5 independent cultures, * p < 0.05 vs. control, # p < 0.05 vs. oxLDL group.
3.3. Role of the Redox-sensitive ERK1/2 Pathway in oxLDL-stimulated ET-1 Gene Expression
Early evidence noted an essential role for stress signaling activation in response to oxLDL challenge [34]. Data presented in Figure 3A displayed that oxLDL challenge overtly promoted ERK1/2 phosphorylation without affecting pan protein expression of ERK1/2, the effect of which was nullified by NAC or the MEK inhibitor PD98059 (p < 0.0001). Neither NAC nor PD98059 elicited any notable effects on ERK1/2 activation by themselves. To discern the role of ERK1/2 signaling in oxLDL-evoked ET-1 signaling, HUVECs were challenged with oxLDL (35 μg/mL) in the absence or presence of NAC or PD98059 prior to assessment of ET-1 secretion and promotor activity. Our results showed that oxLDL promoted secretion and promoter activity of ET-1, the effects of which were nullified by NAC and PD98059 with little effect from NAC or PD98059 themselves (p < 0.0001, Figure 3B—C). These data favor an obligatory role for ERK1/2 activation in oxLDL-fostered ET-1 gene expression.

Figure 3. ERK activation, ET-1 secretion and ET-1 promotor activity in HUVECs challenged with oxLDL (35 µg/mL) for 24 h in the absence or presence of the antioxidant NAC (10 µM) or the MEK inhibitor PD98059 (30 µM). A: pERK-to-ERK ratio, inset: Representative gel blots depicting levels of pan and phosphorylated ERK; B: ET-1 secretion; and C: ET-1 promotor activity. Mean ± SEM, n = 5–7 independent cultures, * p < 0.05 vs. control, # p < 0.05 vs. oxLDL group.
3.4. Identification of oxLDL-responsive Regulatory Elements
To discern the nucleotide sequences responsible for the regulation of oxLDL-induced ET-1 gene expression, a series of deletion constructs was transfected into HUVECs. Fusion plasmids with truncated length of 5’-flanking sequence of ET-1 from –566 bpLuc to -250 bpLuc retained oxLDL(35 μg/mL)-evoked elevation in luciferase activity following 24 h of incubation. Intriguingly, fusion plasmid from –233 and –185 bpLuc significantly dampened luciferase activity in both basal and oxLDL-challenged conditions (p < 0.0001, Figure 4), denoting AP-1 binding sites upstream of 250 bp as possible transcription initiation site for oxLDL-induced ET-1 expression.

Figure 4. ET-1 promoter activity in oxLDL (35 µg/mL, 24 h)-treated HUVECs transiently challenged with a luciferase reporter construct pGL3-ET-1-431/+135, containing 566 bp from -431 to +135 of ET-1 promoter. HUVECs were co-transfected with pSV-β-gal for correction of transfection efficiency. A: Representative scheme depicting segmental truncation of the luciferase reporter construct pGL3-ET-1-431/+135; The position of cis-acting elements of NF-1, GATA-2, AP-1, CAAT and TATA are illustrated in the 5’-upstream of ET-1 gene; B: ET-1 promotor activity in HUVECs co-transfected with pSV-β-galactosidase plasmid and one of the following deletion constructs: -566 bp, –428 bp, –284 bp, –250 bp, –233 bp, –185 bp. HUVECs were transfected for 48 h prior to oxLDL challenge for another 24 h. ET-1 promoter/enhancer activity presented as the ratio of luciferase activity/β-galactosidase. Mean ± SEM, n = 5, * p < 0.05 vs. control group.
3.5. Crucial Role of AP-1 Motif in oxLDL-induced Increases in ET-1 Promoter Activity
The ET-1 promoter contains multiple AP-1 binding sites under the governance of various signaling pathways [35,36]. In particular, the AP-1 binding site functions as an essential cis element in ROS-evoked ET-1 gene expression [37]. Our results indicated that oxLDL (35 μg/mL) overtly upregulated AP-1 protein expression and its reporter activity in endothelial cells, the effect of which was annihilated by NAC or PD98059 with little effect from NAC or PD98059 themselves (p = 0.005 for panel A and p < 0.0001 for panel B, Figure 5A—B). In addition, transfection with a reporter construct –250 Luc, containing a 2 bp mutation at the AP-1 site, nullified both basal and oxLDL-stimulated AP-1 promoter activity (p < 0.0001, Figure 5C). These observations favor an essential role for ROS and ERK1/2 in oxLDL-evoked transcriptional activity of AP-1. Our data also denote a crucial role for the AP-1 binding element in oxLDL-induced ET-1 gene expression.

Figure 5. Role of AP-1 motif in oxLDL-induced increases in ET-1 promoter activity. A: AP-1 protein levels. Inset: Representative gel blots depicting levels of AP-1 in response to oxLDL challenge in the absence or presence of NAC (10 µM) or PD98059 (30 µM); B: Basal and oxLDL-induced AP-1 promotor activity in HUVECs transfected with native (250 LUC) or mutated (mut) AP-1. Luciferase activity was measured in oxLDL (35 µg/mL, 24 h)-treated HUVECs transfected with the ET-1 promoter deletion constructs. Mean ± SEM, n = 5. * p < 0.05 vs. control.
4. Discussion
The salient findings from our study revealed that oxLDL fosters mRNA expression and release of ET-1 in endothelial cells through generation of ROS as a second messenger to activate ERK1/2 signaling and possibly apoptosis. Compelling earlier evidence has indicated that ROS in particular O2- is capable of coupling to the initiation codon on ET-1 gene to turn on ET-1 transcription [10,23]. This coincides with our current finding where antioxidant NAC reversed oxLDL-evoked mRNA expression, secretion and promoter activity of ET-1, in conjunction with reversal of oxLDL-evoked ROS accumulation and apoptosis in HUVECs. Collectively, our findings support an important role for redox signaling in oxLDL-evoked ET-1 expression in endothelial cells.
In our study, functional GO and GSEA annotation analyses of DEGs were performed using gene expression datasets of oxLDL-challenged endothelial cells. Network plot of biological function and KEGG enrichment were employed for visualization and interpretation of pathway enrichment analysis. First, the GO terms (e.g., cell division, cell proliferation) and KEGG pathways (e.g., cell cycle, p53 signaling, DNA replication, cancer-related pathways) validated the enrichment analysis results and supported our experimental finding in MAPK activation, ROS accumulation, apoptosis and ET-1 signaling in ox-LDL-evoked endothelial dysfunction. ROS is commonly known to turn on multiple genes to govern a wide array of pathophysiological processes including migration, proliferation, inflammation, and various forms of cell death [10,23]. Numerous proinflammatory instigators such as oxLDL and ET-1 are capable of imposing unfavorable pathophysiological effects through ROS accumulation [8,38,39]. Among various stress signaling cascades, MAPK appears to be a predominant one involved in signaling transduction where ERK1/2 serves as a cardinal member. In response to ROS production, MAPK is turned on and becomes translocated into the nucleus to activate various transcriptional factors to govern gene expression [40]. Likewise, our current data noted activation of ERK1/2 in response to oxLDL challenge. Moreover, the specific MEK inhibitor PD98059 abrogates oxLDL-evoked ET-1 expression in endothelial cells, somewhat consistent with earlier notion [37]. These results favor an essential role for ERK1/2 signaling in ET-1 genetic regulation in the face of oxLDL challenge. In our hands, PD98059 effectively suppressed oxLDL-evoked ERK1/2 phosphorylation, reminiscent of NAC, further supporting the essential role for redox signaling in ERK1/2 activation. Our data also noted that PD98059 alleviated oxLDL-triggered mRNA expression, secretion, and promoter activity of ET-1 in endothelial cells, favoring a role of MAPK in ET-1 induction.
Our data noted that inhibition of ROS and ERK1/2 using pharmacological inhibitors effectively nullified oxLDL-evoked ET-1 promotor activity. Human ET-1 gene codon is comprised of 12-O-tetradecanoylphorbol 13-acetate (TPA) response elements (TRE) which are perceived as the binding site for the transcriptional factor AP-1. Transcription factor AP-1 contains either a Jun-Jun homodimer or a Jun-Fos heterodimer and is implicated in redox balance, ROS, growth factors, cytokine release, angiogenesis, vascular development and endothelial function. Levels of AP-1 appear to be transiently induced in endothelial cells upon stress [41]. Once activated, AP-1 dimer binds to DNA sequences within the regulatory domains of mitogen-responsive genes TRE in the promoter regions of target gene to exert its function. Human ET-1 promoter encompasses an AP-1 domain responsive to ROS challenge. This is in line with the notion of ROS as a vivid second messenger for AP-1 activation. In our current study, oxLDL was shown to activate AP-1 and AP-1 luciferase activity, consistent with its ability of MAPK activation to foster AP-1 expression and DNA binding [34,42]. Meanwhile, antioxidants and free radical scavengers may suppress the DNA binding affinity and transcriptional capacity of AP-1 [43,44], further consolidate the essential role of redox signaling in AP-1 function. Our results noted that oxLDL overtly upregulated c-jun/AP-1 protein and AP-1 reporter gene function, in a ROS- and ERK1/2-dependent manner. Changes in luciferase reporter genes and ET-1 gene promoter also denoted an essential role for the AP-1 sites in ET-1 promoter function in endothelial cells. Moreover, the AP-1 binding sequence serves as an important cis element for oxLDL-evoked ET-1 gene expression. These findings demonstrated a rather critical role for the redox-sensitive transcription factor AP-1 in oxLDL-induced regulation of ET-1 gene expression. Our data suggested that oxLDL evoked ET-1 gene expression, at least in part, via an ERK1/2-dependent pathway in vascular endothelial cells.
Experimental limitations: Several limitations prevail for our current study. First, the levels of ET-1 are overtly elevated in many cardiovascular diseases including atherosclerosis [5,6]; oxLDL may not be the only instigator in real clinical settings. Caution should be taken in translating our current experimental findings in the realm of atherosclerosis and other vascular diseases. Second, other signaling mechanisms such as proinflammatory cascades may also participate in oxLDL-evoked regulation of ET-1 signaling. Although it is beyond the scope of the current study, these potential "stakeholders" should not be discounted.
5. Conclusion
In summary, findings from our study suggested that oxLDL provoked activation of ERK1/2 through ROS accumulation. Next, transcriptional factor AP-1 and ET-1 expression were turned on, contributing to oxLDL-induced endothelial dysfunction and atherosclerosis. Involvement of ROS in oxLDL-induced ERK1/2 activation received further support where NAC effectively suppressed ERK1/2 phosphorylation, AP-1 activation as well as gene expression and protein secretion of ET-1. These findings added additional support for the earlier finding that oxLDL regulates expression and secretion of ET-1 in endothelial cells by way of governance of intracellular Ca2+ recruitment, PKC and MAPK [22]. Nonetheless, further study is still warranted to identify ET-1/AP-1-targeted therapy in endothelial dysfunction, which would benefit the ultimate outcomes of atherosclerosis and inflammation.
Author Contributions: HX, JD, JT, WW data collection and analysis; YW: helpful discussion; SD and JR manuscript drafting, editing and supervision of the study.
Funding: This work was supported in part by the National Natural Sciences Foundation of China (30170376, 30700293).
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: Not applicable.
Acknowledgments: We thank Professors Chengyu Jiang, Youhe Gao, Xiaodong Zhang, Xiaoming Wang and Baosheng Qi from Peking Union Medical College for their generous support for this project. Technical assistance from Ms. Xiaomei
Zhou and Ms. Sucan Ma from the Institute of Basic Medical Sciences, Chinese Academy of Medical Science is greatly appreciated.
Conflicts of Interest: The authors declare that they have no conflict of interest.
Publication Ethics: Authors should conform to the publication ethics. No human subject or animals were involved.
References
- Chang X.; Lochner A.; Wang H.H.; et al. Coronary microvascular injury in myocardial infarction: perception and knowledge for mitochondrial quality control. Theranostics, 2021, 11(14): 6766-6785. DOI: https://doi.org/10.7150/thno.60143
- de Oliveira M.G.; Nadruz W., Jr.; Mónica F.Z. Endothelial and vascular smooth muscle dysfunction in hypertension. Biochem. Pharmacol., 2022, 205: 115263. DOI: https://doi.org/10.1016/j.bcp.2022.115263
- Ren J.; Wu N.N.; Wang S.Y.; et al. Obesity cardiomyopathy: evidence, mechanisms, and therapeutic implications. Physiol. Rev., 2021, 101(4): 1745-1807. DOI: https://doi.org/10.1152/physrev.00030.2020
- Ren J.; Wei C.M. New sniper assignment for a celebrity—role of endothelin-1 in diabetic cardiomyopathy. J. Cardiothoracic-Renal Res., 2006, 1(1): 30-32. DOI: https://doi.org/10.1016/j.jccr.2005.11.006
- Kostov K. The causal relationship between endothelin-1 and hypertension: focusing on endothelial dysfunction, arterial stiffness, vascular remodeling, and blood pressure regulation. Life, 2021, 11(9): 986. DOI: https://doi.org/10.3390/life11090986
- Barton M.; Yanagisawa M. Endothelin: 30 years from discovery to therapy. Hypertension, 2019, 74(6): 1232-1265. DOI: https://doi.org/10.1161/HYPERTENSIONAHA.119.12105
- Eroglu E.; Kocyigit I.; Lindholm B. The endothelin system as target for therapeutic interventions in cardiovascular and renal disease. Clin. Chim. Acta, 2020, 506: 92-106. DOI: https://doi.org/10.1016/j.cca.2020.03.008
- Han Q.A.; Yan C.H.; Wang L.F.; et al. Urolithin A attenuates ox-LDL-induced endothelial dysfunction partly by modulating microRNA-27 and ERK/PPAR-γ pathway. Mol. Nutr. Food Res., 2016, 60(9): 1933-1943. DOI: https://doi.org/10.1002/mnfr.201500827
- Yang M.Y.; Wang Y.B.; Han B.; et al. Activation of aldehyde dehydrogenase 2 slows down the progression of atherosclerosis via attenuation of ER stress and apoptosis in smooth muscle cells. Acta Pharmacol. Sin., 2018, 39(1): 48-58. DOI: https://doi.org/10.1038/aps.2017.81
- Engin A. Endothelial dysfunction in obesity. Adv. Exp. Med. Biol., 2017, 960: 345-379. DOI: https://doi.org/10.1007/978-3-319-48382-5_15
- Xu H.S.; Duan J.H.; Dai S.L.; et al. α-Zearalanol attenuates oxLDL-induced ET-1 gene expression, ET-1 secretion and redox-sensitive intracellular signaling activation in human umbilical vein endothelial cells. Toxicol. Lett., 2008, 179(3): 163-168. DOI: https://doi.org/10.1016/j.toxlet.2008.05.005
- Xu H.S.; Duan J.H.; Dai S.L.; et al. Phytoestrogen α-zearalanol antagonizes oxidized LDL-induced inhibition of nitric oxide production and stimulation of endothelin-1 release in human umbilical vein endothelial cells. Endocrine, 2004, 25(3): 235-245. DOI: https://doi.org/10.1385/ENDO:25:3:235
- Zhao J.; Zhang Q.; Liu J.; et al. Effect of endomorphins on HUVECs treated by ox-LDL and its related mechanisms. J. Diabetes Res., 2016, 2016: 9741483. DOI: https://doi.org/10.1155/2016/9741483
- Ren J.; Pulakat L.; Whaley-Connell A.; et al. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med., 2010, 88(10): 993-1001. DOI: https://doi.org/10.1007/s00109-010-0663-9
- Hu N.; Ren J. Reactive oxygen species regulate myocardial mitochondria through post-translational modification. React. Oxygen Species, 2016, 2(4): 264-271. DOI: https://doi.org/10.20455/ros.2016.845
- Wang S.Y.; Guo W.; Ren J. Stress signaling in paraquat-induced target organ toxicity. React. Oxygen Species, 2016, 1(2): 131-140. DOI: https://doi.org/10.20455/ros.2016.827
- Panzhinskiy E.; Ren J.; Nair S. Protein tyrosine phosphatase 1B and insulin resistance: role of endoplasmic reticulum stress/reactive oxygen species/nuclear factor kappa B axis. PLoS One, 2013, 8(10): e77228. DOI: https://doi.org/10.1371/journal.pone.0077228
- Roe N.D.; Ren J. Nitric oxide synthase uncoupling: a therapeutic target in cardiovascular diseases. Vasc. Pharmacol., 2012, 57(5/6): 168-172. DOI: https://doi.org/10.1016/j.vph.2012.02.004
- Wold L.E.; Ceylan-Isik A.F.; Ren J. Oxidative stress and stress signaling: menace of diabetic cardiomyopathy. Acta Pharmacol. Sin., 2005, 26(8): 908-917. DOI: https://doi.org/10.1111/j.1745-7254.2005.00146.x
- Liu Q.W.; Han L.M.; Du Q.F.; et al. The association between oxidative stress, activator protein-1, inflammatory, total antioxidant status and artery stiffness and the efficacy of olmesartan in elderly patients with mild-to-moderate essential hypertension. Clin. Exp. Hypertens., 2016, 38(4): 365-369. DOI: https://doi.org/10.3109/10641963.2015.1131285
- Lin S.J.; Shyue S.K.; Liu P.L.; et al. Adenovirus-mediated overexpression of catalase attenuates oxLDL-induced apoptosis in human aortic endothelial cells via AP-1 and C-Jun N-terminal kinase/extracellular signal-regulated kinase mitogen-activated protein kinase pathways. J. Mol. Cell. Cardiol., 2004, 36(1): 129-139. DOI: https://doi.org/10.1016/j.yjmcc.2003.10.011
- Cheng T.H.; Cheng P.Y.; Shih N.L.; et al. Involvement of reactive oxygen species in angiotensin Ⅱ-induced endothelin-1 gene expression in rat cardiac fibroblasts. J. Mol. Cell. Cardiol., 2003, 42(10): 1845-1854. DOI: https://doi.org/10.1016/j.jacc.2003.06.010
- Prieto D.; Contreras C.; Sánchez A. Endothelial dysfunction, obesity and insulin resistance. Curr. Vasc. Pharmacol., 2014, 12(3): 412-426. DOI: https://doi.org/10.2174/1570161112666140423221008
- Dong F.; Zhang X.C.; Wold L.E.; et al. Endothelin-1 enhances oxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: role of ETB receptor, NADPH oxidase and caveolin-1. Br. J. Pharmacol., 2005, 145(3): 323-333. DOI: https://doi.org/10.1038/sj.bjp.0706193
- Ritchie M.E.; Phipson B.; WuD.; et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res., 2015, 43(7): e47. DOI: https://doi.org/10.1093/nar/gkv007
- Yu G.C.; Wang L.G.; Han Y.Y.; et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS, 2012, 16(5): 284-287. DOI: https://doi.org/10.1089/omi.2011.0118
- Kanehisa M.; Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res., 2000, 28(1): 27-30. DOI: https://doi.org/10.1093/nar/28.1.27
- Walter W.; Sánchez-Cabo F.; Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics, 2015, 31(17): 2912-2914. DOI: https://doi.org/10.1093/bioinformatics/btv300
- Subramanian A.; Tamayo P.; Mootha V.K.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A., 2005, 102(43): 15545-15550. DOI: https://doi.org/10.1073/pnas.0506580102
- Xu H.S.; Duan J.H.; Wang W.; et al. Reactive oxygen species mediate oxidized low-density lipoprotein-induced endothelin-1 gene expression via extracellular signal-regulated kinase in vascular endothelial cells. J. Hypertens., 2008, 26(5): 956-963. DOI: https://doi.org/10.1097/HJH.0b013e3282f56bb7
- Min J.; Wu L.; Liu Y.D.; et al. Empagliflozin attenuates trastuzumab-induced cardiotoxicity through suppression of DNA damage and ferroptosis. Life Sci., 2023, 312: 121207. DOI: https://doi.org/10.1016/j.lfs.2022.121207
- Ceylan-Isik A.F.; Dong M.L.; Zhang Y.M.; et al. Cardiomyocyte-specific deletion of endothelin receptor A rescues aging-associated cardiac hypertrophy and contractile dysfunction: role of autophagy. Basic Res. Cardiol., 2013, 108(2): 335. DOI: https://doi.org/10.1007/s00395-013-0335-3
- Duan J.H.; Dai S.L.; Fang C.X.; et al. Phytoestrogen α-zearalanol antagonizes homocysteine-induced imbalance of nitric oxide/endothelin-1 and apoptosis in human umbilical vein endothelial cells. Cell Biochem. Biophys., 2006, 45(2): 137-145. DOI: https://doi.org/10.1385/CBB:45:2:137
- Cho S.; Hazama M.; Urata Y.; et al. Protective role of glutathione synthesis in response to oxidized low density lipoprotein in human vascular endothelial cells. Free Radical Biol. Med., 1999, 26(5/6): 589-602. DOI: https://doi.org/10.1016/S0891-5849(98)00232-9
- Delerive P.; Martin-Nizard F.; Chinetti G.; et al. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ. Res., 1999, 85(5): 394-402. DOI: https://doi.org/10.1161/01.RES.85.5.394
- Yamashita K.; Discher D.J.; Hu J.; et al. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J. Biol. Chem., 2001, 276(16): 12645-12653. DOI: https://doi.org/10.1074/jbc.M011344200
- Hsu Y.H.; Chen J.J.; Chang N.C.; et al. Role of reactive oxygen species-sensitive extracellular signal-regulated kinase pathway in angiotensin Ⅱ-induced endothelin-1 gene expression in vascular endothelial cells. J. Vasc. Res., 2004, 41(1): 64-74. DOI: https://doi.org/10.1159/000076247
- Fang C.; Yang Z.C.; Shi L.; et al. Circulating sestrin levels are increased in hypertension patients. Dis. Markers, 2020, 2020: 3787295. DOI: https://doi.org/10.1155/2020/3787295
- Tian H.G.; Li S.Z.; Yu K.H. DJ‑1 alleviates high glucose‑induced endothelial cells injury via PI3K/Akt‑eNOS signaling pathway. Mol. Med. Rep., 2018, 17(1): 1205-1211. DOI: https://doi.org/10.3892/mmr.2017.7975
- Arif A.; Alameri A.A.; Tariq U.B.; et al. The functions and molecular mechanisms of Tribbles homolog 3 (TRIB3) implicated in the pathophysiology of cancer. Int. Immunopharmacol., 2023, 114: 109581. DOI: https://doi.org/10.1016/j.intimp.2022.109581
- Yoshitomi Y.; Ikeda T.; Saito-Takatsuji H.; et al. Emerging role of AP-1 transcription factor JunB in angiogenesis and vascular development. Int. J. Mol. Sci., 2021, 22(6): 2804. DOI: https://doi.org/10.3390/ijms22062804
- Estruch M.; Sanchez-Quesada J.L.; Ordoñez-Llanos J.; et al. Inflammatory intracellular pathways activated by electronegative LDL in monocytes. Biochim. Biophys. Acta, 2016, 1861(9 Pt A): 963-969. DOI: https://doi.org/10.1016/j.bbalip.2016.05.010
- Feng R.T.; Bowman L.L.; Lu Y.J.; et al. Blackberry extracts inhibit activating protein 1 activation and cell transformation by perturbing the mitogenic signaling pathway. Nutr. Cancer, 2004, 50(1): 80-89. DOI: https://doi.org/10.1207/s15327914nc5001_11
- Maggi-Capeyron M.F.; Ceballos P.; Cristol J.P.; et al. Wine phenolic antioxidants inhibit AP-1 transcriptional activity. J. Agric. Food Chem., 2001, 49(11): 5646-5652. DOI: https://doi.org/10.1021/jf010595x


